Translating Translation to Mechanisms of Cardiac Hypertrophy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Global Regulators of Translation
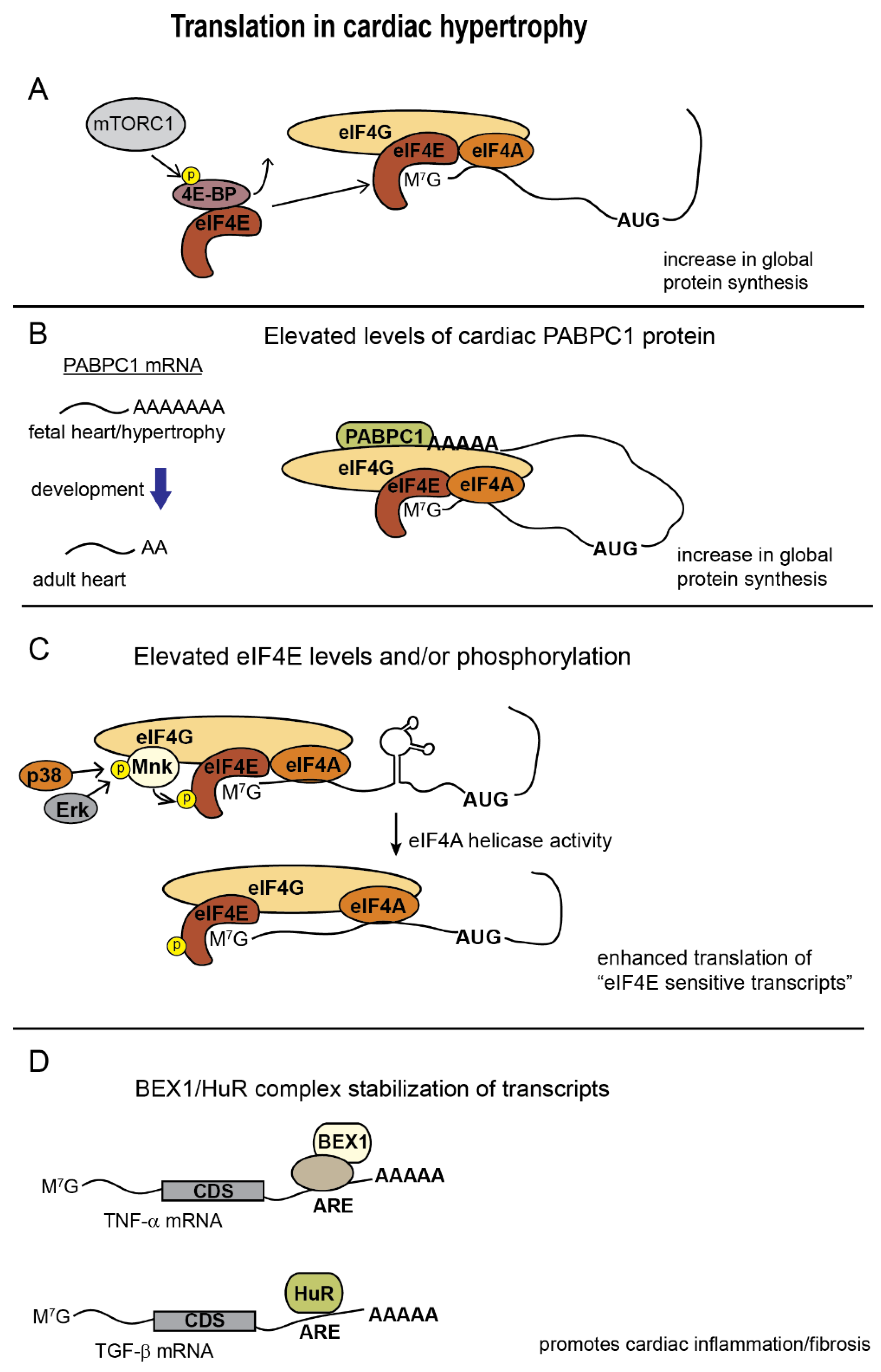
2.1. eIF4E
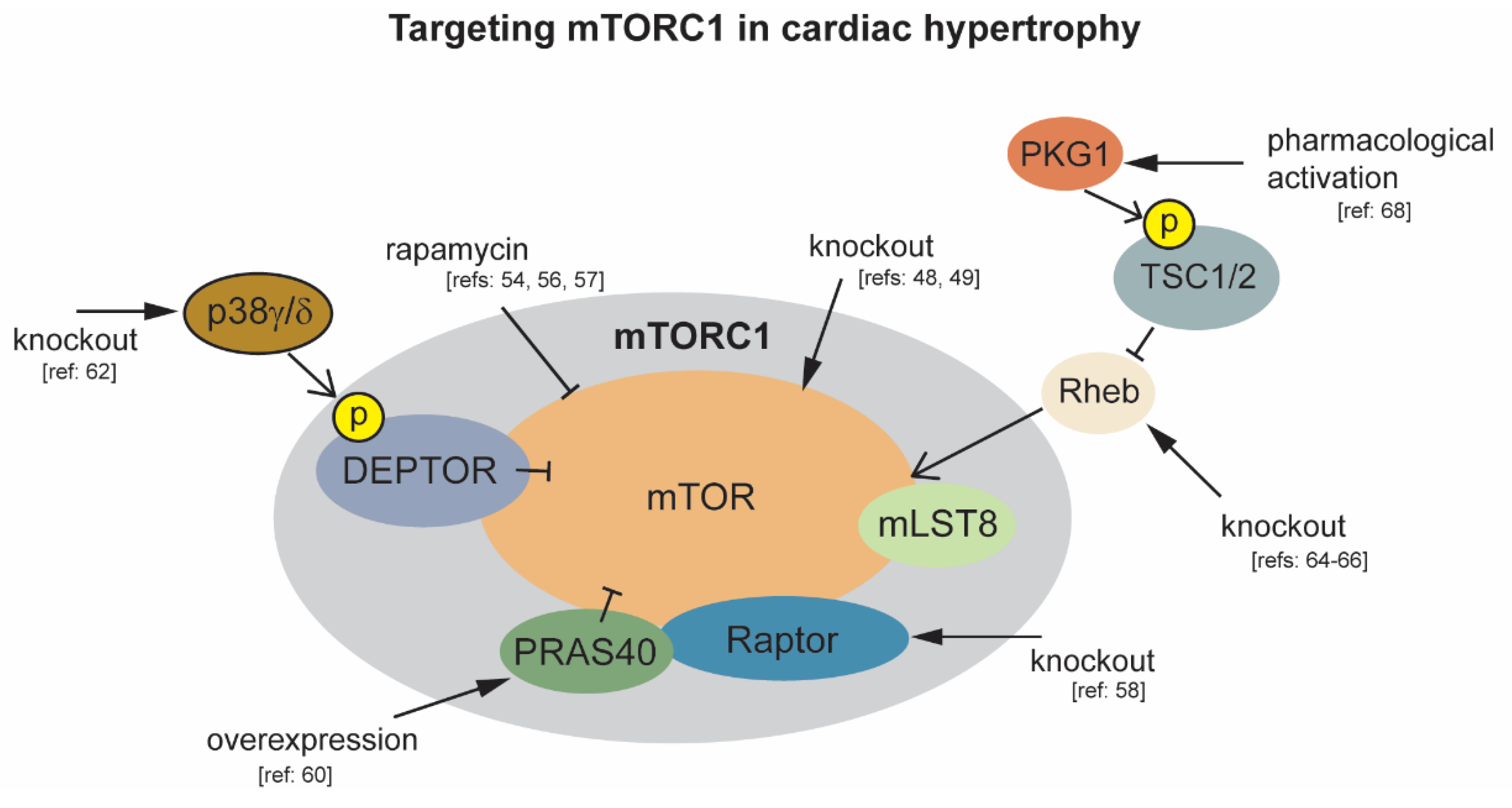
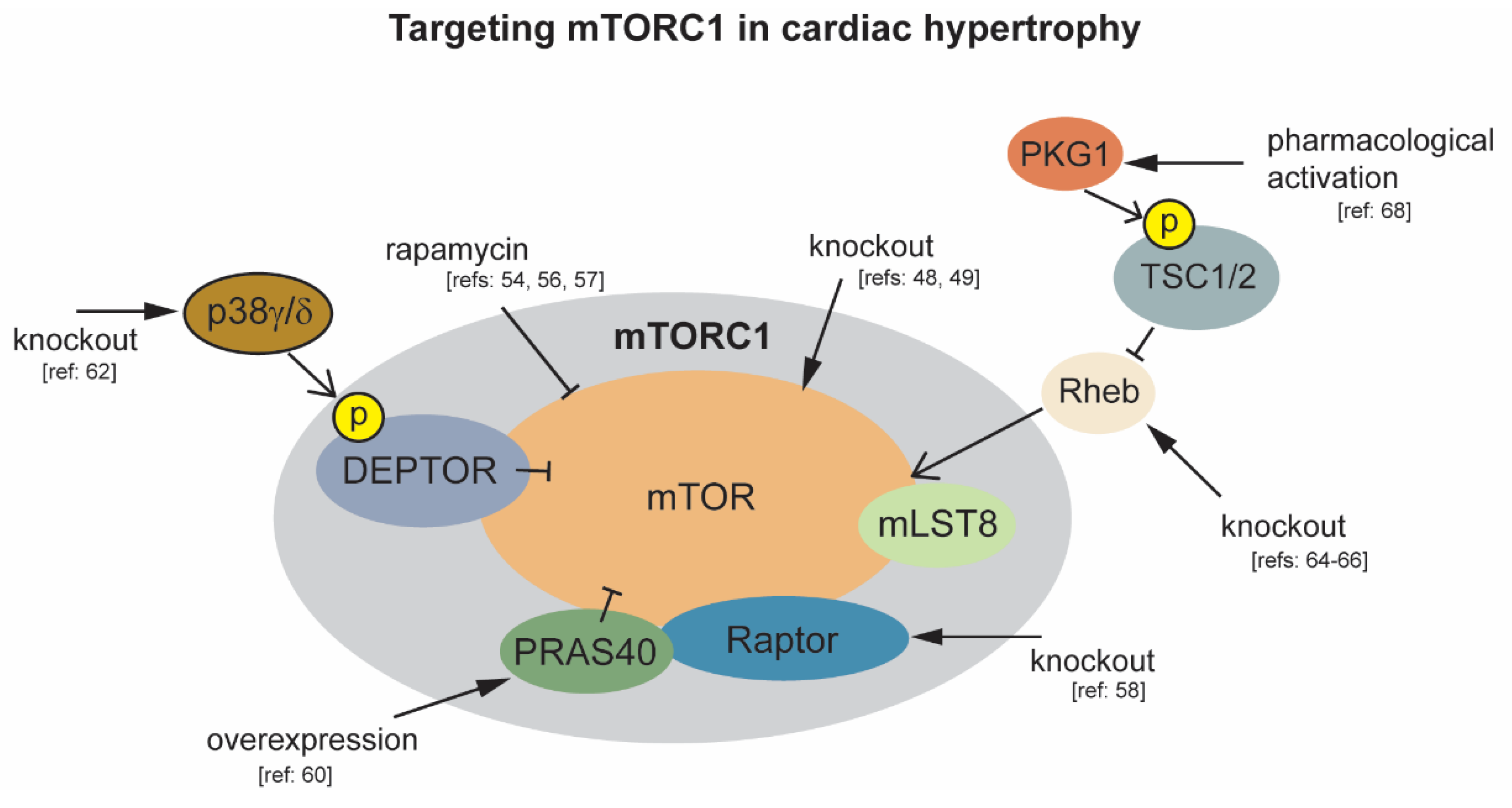
2.2. mTORC1
2.3. Endoplasmic Reticulum Stress and the Unfolded Protein Response
2.4. PABPC1
3. Local Regulatory Elements
AU-Rich Element Binding Proteins
4. Alternative Mechanisms of Translation Initiation in Cardiac Biology
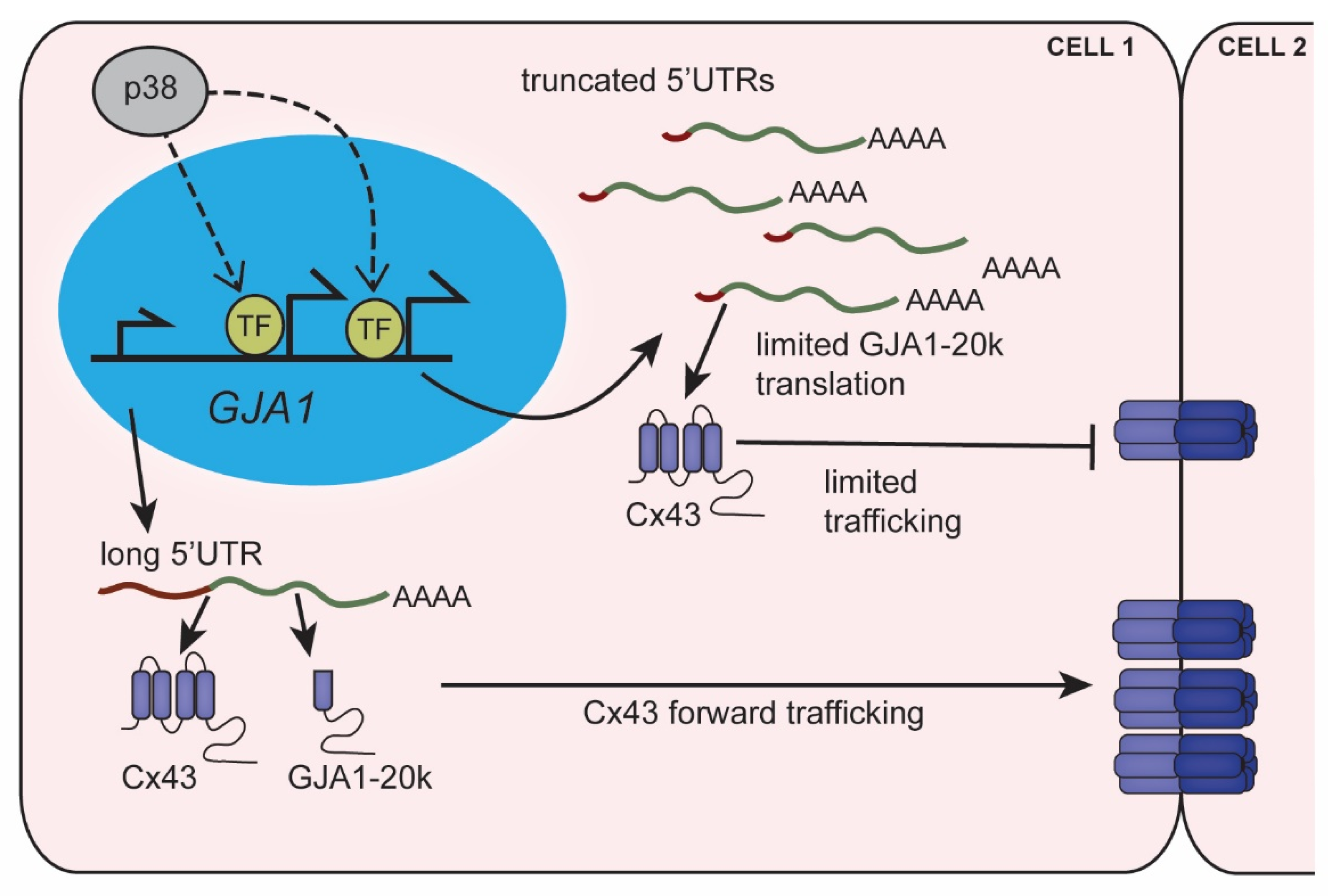
4.1. Altered Connexin43 Protein Translation in Heart Disease
4.2. Translation as a Regulator of Cardiac Ion Channel Function
5. Future Perspectives
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Petersen, R.; Baserga, R. Nucleic acid and protein synthesis in cardiac muscle of growing and adult mice. Exp. Cell Res. 1965, 40, 340–352. [Google Scholar] [PubMed]
- Haider, A.W.; Larson, M.G.; Benjamin, E.J.; Levy, D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 1998, 32, 1454–1459. [Google Scholar] [PubMed] [Green Version]
- Perrino, C.; Prasad, S.V.N.; Mao, L.; Noma, T.; Yan, Z.; Kim, H.-S.; Smithies, O.; Rockman, H.A. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J. Clin. Investig. 2006, 116, 1547–1560. [Google Scholar] [PubMed] [Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar]
- Schwanhäusser, B.; Busse, R.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar]
- Van Heesch, S.; Witte, F.; Schneider-Lunitz, V.; Schulz, J.F.; Adami, E.; Faber, A.B.; Kirchner, M.; Maatz, H.; Blachut, S.; Sandmann, C.-L.; et al. The Translational Landscape of the Human Heart. Cell 2019, 178, 242–260.e29. [Google Scholar]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136, 731–745. [Google Scholar]
- Archer, S.; Shirokikh, N.E.; Beilharz, T.H.; Preiss, T. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature 2016, 535, 570–574. [Google Scholar]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar]
- Wells, S.E.; Hillner, P.E.; Vale, R.D.; Sachs, A.B. Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell 1998, 2, 135–140. [Google Scholar]
- Kozak, M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 1986, 44, 283–292. [Google Scholar] [PubMed]
- Hiremath, L.S.; Webb, N.R.; Rhoads, R.E. Immunological detection of the messenger RNA cap-binding protein. J. Biol. Chem. 1985, 260, 7843–7849. [Google Scholar]
- Duncan, R.; Milburn, S.C.; Hershey, J.W. Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF-4F suggest a role in translational control. Heat shock effects on eIF-4F. J. Biol. Chem. 1987, 262, 380–388. [Google Scholar]
- Rousseau, D.; Kaspar, R.; Rosenwald, I.; Gehrke, L.; Sonenberg, N. Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proc. Natl. Acad. Sci. USA 1996, 93, 1065–1070. [Google Scholar] [PubMed] [Green Version]
- Pelletier, J.; Sonenberg, N. Insertion mutagenesis to increase secondary structure within the 5′ noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell 1985, 40, 515–526. [Google Scholar] [PubMed]
- Koromilas, A.; Lazaris-Karatzas, A.; Sonenberg, N. mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 1992, 11, 4153–4158. [Google Scholar]
- Feoktistova, K.; Tuvshintogs, E.; Do, A.; Fraser, C.S. Human eIF4E promotes mRNA restructuring by stimulating eIF4A helicase activity. Proc. Natl. Acad. Sci. USA 2013, 110, 13339–13344. [Google Scholar]
- Svitkin, Y.V.; Pause, A.; Haghighat, A.; Pyronnet, S.; Witherell, G.; Belsham, G.J.; Sonenberg, N. The requirement for eukaryotic initiation factor 4A (eIF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 2001, 7, 382–394. [Google Scholar]
- Truitt, M.L.; Conn, C.S.; Shi, Z.; Pang, X.; Tokuyasu, T.; Coady, A.M.; Seo, Y.; Barna, M.; Ruggero, D. Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell 2015, 162, 59–71. [Google Scholar]
- Makhlouf, A.A.; McDermott, P.J. Increased expression of eukaryotic initiation factor 4E during growth of neonatal rat cardiocytes in vitro. Am. J. Physiol. Content 1998, 274, H2133–H2142. [Google Scholar]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, B.; Cai, A.-L.; Keiper, B.D.; Minich, W.B.; Méndez, R.; Beach, C.M.; Stepinski, J.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Phosphorylation of Eukaryotic Protein Synthesis Initiation Factor 4E at Ser-209. J. Biol. Chem. 1995, 270, 14597–14603. [Google Scholar]
- Ueda, T.; Watanabe-Fukunaga, R.; Fukuyama, H.; Nagata, S.; Fukunaga, R. Mnk2 and Mnk1 Are Essential for Constitutive and Inducible Phosphorylation of Eukaryotic Initiation Factor 4E but Not for Cell Growth or Development. Mol. Cell. Biol. 2004, 24, 6539–6549. [Google Scholar]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.; Cooper, J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar]
- Korneeva, N.L.; Song, A.; Gram, H.; Edens, M.A.; Rhoads, R.E. Inhibition of Mitogen-activated Protein Kinase (MAPK)-interacting Kinase (MNK) Preferentially Affects Translation of mRNAs Containing Both a 5′-Terminal Cap and Hairpin*. J. Biol. Chem. 2015, 291, 3455–3467. [Google Scholar]
- Singh, K.; Silva, R.L.; Malina, A.; Mills, J.R.; Zhu, H.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Teruya-Feldstein, J.; Pelletier, J.; et al. Dissecting eIF4E action in tumorigenesis. Genome Res. 2007, 21, 3232–3237. [Google Scholar]
- Chorghade, S.; Seimetz, J.; Emmons, R.; Yang, J.; Bresson, S.; De Lisio, M.; Parise, G.; Conrad, N.K.; Kalsotra, A. Poly(A) tail length regulates PABPC1 expression to tune translation in the heart. eLife 2017. [Google Scholar] [CrossRef]
- Spruill, L.S.; Baicu, C.F.; Zile, M.R.; McDermott, P.J. Selective translation of mRNAs in the left ventricular myocardium of the mouse in response to acute pressure overload. J. Mol. Cell. Cardiol. 2007, 44, 69–75. [Google Scholar]
- Siehl, D.; Chua, B.H.; Lautensack-Belser, N.; Morgan, H.E. Faster protein and ribosome synthesis in thyroxine-induced hypertrophy of rat heart. Am. J. Physiol. Physiol. 1985, 248, C309–C319. [Google Scholar]
- Brandenburger, Y. Increased expression of UBF is a critical determinant for rRNA synthesis and hypertrophic growth of cardiac myocytes. FASEB J. 2001. [Google Scholar] [CrossRef]
- Fan, C.; Iacobas, D.A.; Zhou, D.; Chen, Q.; Lai, J.K.; Gavrialov, O.; Haddad, G.G. Gene expression and phenotypic characterization of mouse heart after chronic constant or intermittent hypoxia. Physiol. Genom. 2005, 22, 292–307. [Google Scholar]
- Nagai, R.; Low, R.B.; Stirewalt, W.S.; Alpert, N.R.; Litten, R.Z. Efficiency and capacity of protein synthesis are increased in pressure overload cardiac hypertrophy. Am. J. Physiol. Circ. Physiol. 1988, 255, H325–H328. [Google Scholar]
- Mezzetti, G. Peptide chain initiation and analysis of in vitro translation products in rat heart undergoing hypertrophic growth. J. Mol. Cell. Cardiol. 1983, 15, 629–635. [Google Scholar] [PubMed]
- Nagatomo, Y.; Carabello, B.A.; Hamawaki, M.; Nemoto, S.; Matsuo, T.; McDermott, P.J. Translational mechanisms accelerate the rate of protein synthesis during canine pressure-overload hypertrophy. Am. J. Physiol. Content 1999, 277, H2176–H2184. [Google Scholar]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [PubMed] [Green Version]
- Kim, D.-H.; Sarbassov, S.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar] [PubMed] [Green Version]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.-I.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell 2002, 110, 177–189. [Google Scholar]
- Haar, E.V.; Lee, S.-I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR Inhibitor Whose Frequent Overexpression in Multiple Myeloma Cells Promotes their Survival. Cell 2009, 137, 873–886. [Google Scholar]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GβL, a Positive Regulator of the Rapamycin-Sensitive Pathway Required for the Nutrient-Sensitive Interaction between Raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar]
- Hara, K.; Yonezawa, K.; Kozlowski, M.T.; Sugimoto, T.; Andrabi, K.I.; Weng, Q.-P.; Kasuga, M.; Nishimoto, I.; Avruch, J. Regulation of eIF-4E BP1 Phosphorylation by mTOR. J. Biol. Chem. 1997, 272, 26457–26463. [Google Scholar]
- Haghighat, A.; Mader, S.; Pause, A.; Sonenberg, N. Repression of cap-dependent translation by 4E-binding protein 1: Competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995, 14, 5701–5709. [Google Scholar] [PubMed]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar]
- Chauvin, C.; Koka, V.; Nouschi, A.; Mieulet, V.; Hoareau-Aveilla, C.; Dreazen, A.; Cagnard, N.; Carpentier, W.; Kiss, T.; Meyuhas, O.; et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2013, 33, 474–483. [Google Scholar] [PubMed]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genome Res. 2002, 16, 1472–1487. [Google Scholar]
- Sciarretta, S.; Zhai, P.; Maejima, Y.; Del Re, M.P.; Nagarajan, N.; Yee, D.; Liu, T.; Magnuson, M.A.; Volpe, M.; Frati, G.; et al. mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep. 2015, 11, 125–136. [Google Scholar]
- Zhang, D.; Contu, R.; Latronico, M.; Zhang, J.; Zhang, J.L.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Investig. 2010, 120, 2805–2816. [Google Scholar]
- Zhu, Y.; Pires, K.M.P.; Whitehead, K.J.; Olsen, C.D.; Wayment, B.; Zhang, Y.; Bugger, H.; Ilkun, O.; Litwin, S.E.; Thomas, G.; et al. Mechanistic Target of Rapamycin (Mtor) Is Essential for Murine Embryonic Heart Development and Growth. PLoS ONE 2013. [Google Scholar] [CrossRef]
- Mazelin, L.; Panthu, B.; Nicot, A.-S.; Belotti, E.; Tintignac, L.; Teixeira, G.; Zhang, Q.; Risson, V.; Baas, M.; Delaune, E.; et al. mTOR inactivation in myocardium from infant mice rapidly leads to dilated cardiomyopathy due to translation defects and p53/JNK-mediated apoptosis. J. Mol. Cell. Cardiol. 2016, 97, 213–225. [Google Scholar]
- Altamirano, F.; Oyarce, C.; Silva, P.; Toyos, M.; Wilson, C.; Lavandero, S.; Uhlén, P.; Estrada, M. Testosterone induces cardiomyocyte hypertrophy through mammalian target of rapamycin complex 1 pathway. J. Endocrinol. 2009, 202, 299–307. [Google Scholar]
- Simm, A.; Schlüter, K.-D.; Diez, C.; Piper, H.M.; Hoppe, J. Activation of p70S6Kinase byβ-adrenoceptor Agonists on Adult Cardiomyocytes. J. Mol. Cell. Cardiol. 1998, 30, 2059–2067. [Google Scholar] [PubMed]
- Soesanto, W.; Lin, H.-Y.; Hu, E.; Lefler, S.; Litwin, S.E.; Sena, S.; Abel, E.D.; Symons, J.D.; Jalili, T. Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension 2009, 54, 1321–1327. [Google Scholar] [PubMed] [Green Version]
- Sadoshima, J.; Izumo, S. Rapamycin Selectively Inhibits Angiotensin II–Induced Increase in Protein Synthesis in Cardiac Myocytes In Vitro. Circ. Res. 1995, 77, 1040–1052. [Google Scholar] [PubMed]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR Signaling With Rapamycin Regresses Established Cardiac Hypertrophy Induced by Pressure Overload. Circulation 2004, 109, 3050–3055. [Google Scholar]
- Kemi, O.J.; Ceci, M.; Wisløff, U.; Grimaldi, S.; Gallo, P.; Smith, G.L.; Condorelli, G.; Ellingsen, O. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J. Cell. Physiol. 2007, 214, 316–321. [Google Scholar]
- Shioi, T.; McMullen, J.R.; Tarnavski, O.; Converso, K.; Sherwood, M.C.; Manning, W.J.; Izumo, S. Rapamycin Attenuates Load-Induced Cardiac Hypertrophy in Mice. Circulation 2003, 107, 1664–1670. [Google Scholar]
- Choo, A.Y.; Yoon, S.-O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar]
- Shende, P.; Plaisance, I.; Morandi, C.; Pellieux, C.; Berthonneche, C.; Zorzato, F.; Krishnan, J.; Lerch, R.; Hall, M.N.; Rüegg, M.A.; et al. Cardiac Raptor Ablation Impairs Adaptive Hypertrophy, Alters Metabolic Gene Expression, and Causes Heart Failure in Mice. Circulation 2011, 123, 1073–1082. [Google Scholar]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar]
- Völkers, M.; Toko, H.; Doroudgar, S.; Din, S.; Quijada, P.; Joyo, A.Y.; Ornelas, L.; Joyo, E.; Thuerauf, N.J.; Konstandin, M.H.; et al. Pathological hypertrophy amelioration by PRAS40-mediated inhibition of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, 12661–12666. [Google Scholar]
- Gao, D.; Inuzuka, H.; Tan, M.-K.M.; Fukushima, H.; Locasale, J.W.; Liu, P.; Wan, L.; Zhai, B.; Chin, Y.M.R.; Shaik, S.; et al. mTOR Drives Its Own Activation via SCFβTrCP-Dependent Degradation of the mTOR Inhibitor DEPTOR. Mol. Cell 2011, 44, 290–303. [Google Scholar]
- González-Terán, B.; Lopez, J.A.; Rodriguez, E.; Leiva, L.; Martínez-Martínez, S.; Bernal, J.; Jiménez-Borreguero, L.J.; Redondo, J.M.; Vázquez, J.; Sabio, G. p38γ and δ promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation. Nat. Commun. 2016. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Ma, N.; Liu, A.; Shen, X.; Wang, Q.; Liu, Y.; Jiang, Y. Rheb Activates mTOR by Antagonizing Its Endogenous Inhibitor, FKBP38. Science 2007, 318, 977–980. [Google Scholar] [PubMed] [Green Version]
- Tamai, T.; Yamaguchi, O.; Hikoso, S.; Takeda, T.; Taneike, M.; Oka, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Uno, Y.; et al. Rheb (Ras Homologue Enriched in Brain)-dependent Mammalian Target of Rapamycin Complex 1 (mTORC1) Activation Becomes Indispensable for Cardiac Hypertrophic Growth after Early Postnatal Period*. J. Biol. Chem. 2013, 288, 10176–10187. [Google Scholar]
- Cao, Y.; Tao, L.; Shen, S.; Xiao, J.; Wu, H.; Li, B.; Wu, X.; Luo, W.; Xiao, Q.; Hu, X.; et al. Cardiac Ablation of Rheb1 Induces Impaired Heart Growth, Endoplasmic Reticulum-Associated Apoptosis and Heart Failure in Infant Mice. Int. J. Mol. Sci. 2013, 14, 24380–24398. [Google Scholar] [PubMed]
- Wu, X.; Cao, Y.; Nie, J.; Liu, H.; Lu, S.; Hu, X.; Zhu, J.; Zhao, X.; Chen, J.; Chen, X.; et al. Genetic and Pharmacological Inhibition of Rheb1-mTORC1 Signaling Exerts Cardioprotection against Adverse Cardiac Remodeling in Mice. Am. J. Pathol. 2013, 182, 2005–2014. [Google Scholar]
- Garami, A.; Zwartkruis, F.J.T.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar]
- Ranek, M.J.; Kokkonen-Simon, K.; Chen, A.; Dunkerly-Eyring, B.; Pinilla-Vera, M.; Oeing, C.U.; Patel, C.H.; Nakamura, T.; Zhu, G.; Bedja, D.; et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 2019, 566, 264–269. [Google Scholar]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar]
- Fu, H.Y.; Okada, K.-I.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Sawada, T.; Okuda, K.; Asano, Y.; Sanada, S.; et al. Ablation of C/EBP Homologous Protein Attenuates Endoplasmic Reticulum-Mediated Apoptosis and Cardiac Dysfunction Induced by Pressure Overload. Circulation 2010, 122, 361–369. [Google Scholar]
- Okada, K.-I.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged Endoplasmic Reticulum Stress in Hypertrophic and Failing Heart After Aortic Constriction. Circulation 2004, 110, 705–712. [Google Scholar]
- Blackwood, E.A.; Hofmann, C.; Domingo, M.S.; Bilal, A.S.; Sarakki, A.; Stauffer, W.; Arrieta, A.; Thuerauf, N.J.; Kolkhorst, F.W.; Müller, O.; et al. ATF6 Regulates Cardiac Hypertrophy by Transcriptional Induction of the mTORC1 Activator, Rheb. Circ. Res. 2019, 124, 79–93. [Google Scholar] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [PubMed]
- Gallie, D.R. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991, 5, 2108–2116. [Google Scholar] [PubMed] [Green Version]
- Coller, J.; Gray, N.; Wickens, M. mRNA stabilization by poly(A) binding protein is independent of poly(A) and requires translation. Genome Res. 1998, 12, 3226–3235. [Google Scholar]
- Bakheet, T. ARED 3.0: The large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006, 34, 111–114. [Google Scholar]
- Shaw, G.; Kamen, R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 1986, 46, 659–667. [Google Scholar]
- Otsuka, H.; Fukao, A.; Funakami, Y.; Duncan, K.E.; Fujiwara, T. Emerging Evidence of Translational Control by AU-Rich Element-Binding Proteins. Front. Genet. 2019, 10, 332. [Google Scholar]
- Schumacher, S.M.; Prasad, S.V.N. Tumor Necrosis Factor-α in Heart Failure: An Updated Review. Curr. Cardiol. Rep. 2018, 20, 117. [Google Scholar]
- Accornero, F.; Schips, T.G.; Petrosino, J.M.; Gu, S.-Q.; Kanisicak, O.; Van Berlo, J.H.; Molkentin, J.D. BEX1 is an RNA-dependent mediator of cardiomyopathy. Nat. Commun. 2017, 8, 1875. [Google Scholar]
- Green, L.; Anthony, S.R.; Slone, S.; Lanzillotta, L.; Nieman, M.L.; Wu, X.; Robbins, N.; Jones, S.M.; Roy, S.; Owens, A.P.; et al. Human antigen R as a therapeutic target in pathological cardiac hypertrophy. JCI Insight 2019. [Google Scholar] [CrossRef]
- Foltz, G.; Ryu, G.-Y.; Yoon, J.-G.; Nelson, T.; Fahey, J.; Frakes, A.; Lee, H.; Field, L.; Zander, K.; Sibenaller, Z.; et al. Genome-Wide Analysis of Epigenetic Silencing IdentifiesBEX1andBEX2as Candidate Tumor Suppressor Genes in Malignant Glioma. Cancer Res. 2006, 66, 6665–6674. [Google Scholar] [PubMed] [Green Version]
- Vilar, M.; Murillo-Carretero, M.I.; Mira, H.; Magnusson, K.; Besset, V.; Ibáñez, C.F. Bex1, a novel interactor of the p75 neurotrophin receptor, links neurotrophin signaling to the cell cycle. EMBO J. 2006, 25, 1219–1230. [Google Scholar] [PubMed] [Green Version]
- Koo, J.H.; Smiley, M.A.; Lovering, R.M.; Margolis, F.L. Bex1 knock out mice show altered skeletal muscle regeneration. Biochem. Biophys. Res. Commun. 2007, 363, 405–410. [Google Scholar]
- Ma, W.-J.; Cheng, S.; Campbell, C.; Wright, A.; Furneaux, H. Cloning and Characterization of HuR, a Ubiquitously Expressed Elav-like Protein. J. Biol. Chem. 1996, 271, 8144–8151. [Google Scholar]
- Slone, S.; Anthony, S.R.; Wu, X.; Benoit, J.B.; Aube, J.; Xu, L.; Tranter, M. Activation of HuR downstream of p38 MAPK promotes cardiomyocyte hypertrophy. Cell. Signal. 2016, 28, 1735–1741. [Google Scholar] [PubMed] [Green Version]
- Topisirovic, I.; Siddiqui, N.; Orolicki, S.; Skrabanek, L.A.; Tremblay, M.; Hoang, T.; Borden, K.L.B. Stability of Eukaryotic Translation Initiation Factor 4E mRNA Is Regulated by HuR, and This Activity Is Dysregulated in Cancer. Mol. Cell. Biol. 2008, 29, 1152–1162. [Google Scholar]
- James, C.C.; Smyth, J.W. Alternative mechanisms of translation initiation: An emerging dynamic regulator of the proteome in health and disease. Life Sci. 2018, 212, 138–144. [Google Scholar]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar]
- Watatani, Y.; Ichikawa, K.; Nakanishi, N.; Fujimoto, M.; Takeda, H.; Kimura, N.; Hirose, H.; Takahashi, S.; Takahashi, Y. Stress-induced Translation of ATF5 mRNA Is Regulated by the 5′-Untranslated Region. J. Biol. Chem. 2007, 283, 2543–2553. [Google Scholar]
- Lee, Y.-Y.; Cevallos, R.C.; Jan, E. An Upstream Open Reading Frame Regulates Translation of GADD34 during Cellular Stresses That Induce eIF2α Phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar]
- Zeitz, M.J.; Calhoun, P.J.; James, C.C.; Taetzsch, T.; George, K.K.; Robel, S.; Valdez, G.; Smyth, J.W. Dynamic UTR Usage Regulates Alternative Translation to Modulate Gap Junction Formation during Stress and Aging. Cell Rep. 2019. [Google Scholar] [CrossRef] [Green Version]
- Coppen, S.R.; Kaba, R.A.; Halliday, D.; Dupont, E.; Skepper, J.N.; Elneil, S.; Severs, N.J. Comparison of connexin expression patterns in the developing mouse heart and human foetal heart. Mol. Cell. Biochem. 2003, 242, 121–127. [Google Scholar] [PubMed]
- Gutstein, D.E.; Morley, G.E.; Tamaddon, H.; Vaidya, D.; Schneider, M.; Chen, J.; Chien, K.R.; Stuhlmann, H.; Fishman, G.I. Conduction Slowing and Sudden Arrhythmic Death in Mice With Cardiac-Restricted Inactivation of Connexin43. Circ. Res. 2001, 88, 333–339. [Google Scholar] [PubMed] [Green Version]
- A Guerrero, P.; Schuessler, R.B.; Davis, L.M.; Beyer, E.; Johnson, C.M.; A Yamada, K.; E Saffitz, J. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J. Clin. Investig. 1997, 99, 1991–1998. [Google Scholar]
- Lerner, D.L.; Yamada, K.A.; Schuessler, R.B.; E Saffitz, J. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation 2000, 101, 547–552. [Google Scholar]
- Wang, X.; Gerdes, A. Chronic Pressure Overload Cardiac Hypertrophy and Failure in Guinea Pigs: III. Intercalated Disc Remodeling. J. Mol. Cell. Cardiol. 1999, 31, 333–343. [Google Scholar]
- Peters, N.S.; Green, C.; Poole-Wilson, P.A.; Severs, N.J. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation 1993, 88, 864–875. [Google Scholar]
- Kostin, S.; Dammer, S.; Hein, S.; Klövekorn, W.P.; Bauer, E.P.; Schaper, J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc. Res. 2004, 62, 426–436. [Google Scholar]
- Salameh, A.; Krautblatter, S.; Karl, S.; Blanke, K.; Gomez, D.R.; Dhein, S.; Pfeiffer, D.; Janousek, J. The signal transduction cascade regulating the expression of the gap junction protein connexin43 by β-adrenoceptors. Br. J. Pharmacol. 2009, 158, 198–208. [Google Scholar] [PubMed] [Green Version]
- Boulaksil, M.; Bierhuizen, M.F.A.; Engelen, M.A.; Stein, M.; Kok, B.J.M.; Van Amersfoorth, S.C.M.; Vos, M.A.; Van Rijen, H.V.M.; De Bakker, J.M.T.; Van Veen, T.A.B. Spatial Heterogeneity of Cx43 is an Arrhythmogenic Substrate of Polymorphic Ventricular Tachycardias during Compensated Cardiac Hypertrophy in Rats. Front. Cardiovasc. Med. 2016, 3, 1561. [Google Scholar]
- Chang, K.-T.; Cheng, C.-F.; King, P.-C.; Liu, S.-Y.; Wang, G.-S. CELF1 Mediates Connexin 43 mRNA Degradation in Dilated Cardiomyopathy. Circ. Res. 2017, 121, 1140–1152. [Google Scholar] [PubMed] [Green Version]
- Peters, N.S.; Coromilas, J.; Severs, N.J.; Wit, A.L. Disturbed Connexin43 Gap Junction Distribution Correlates With the Location of Reentrant Circuits in the Epicardial Border Zone of Healing Canine Infarcts That Cause Ventricular Tachycardia. Circulation 1997, 95, 988–996. [Google Scholar] [PubMed]
- Smyth, J.; Shaw, R.M. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013, 5, 611–618. [Google Scholar] [PubMed] [Green Version]
- Ul-Hussain, M.; Olk, S.; Schoenebeck, B.; Wasielewski, B.; Meier, C.; Prochnow, N.; May, C.; Galozzi, S.; Marcus, K.; Zoidl, G.; et al. Internal Ribosomal Entry Site (IRES) Activity Generates Endogenous Carboxyl-terminal Domains of Cx43 and Is Responsive to Hypoxic Conditions. J. Biol. Chem. 2014, 289, 20979–20990. [Google Scholar]
- Salat, C.; Sesé, M.; Peula, C.; Cajal, S.R.Y.; Aasen, T. Internal translation of the connexin 43 transcript. Cell Commun. Signal. CCS 2014. [Google Scholar] [CrossRef] [Green Version]
- James, C.C.; Zeitz, M.J.; Calhoun, P.J.; Lamouille, S.; Smyth, J.W. Altered translation initiation of Gja1 limits gap junction formation during epithelial–mesenchymal transition. Mol. Biol. Cell 2018, 29, 797–808. [Google Scholar]
- Basheer, W.A.; Fu, Y.; Shimura, D.; Xiao, S.; Agvanian, S.; Hernandez, D.M.; Hitzeman, T.C.; Hong, T.; Shaw, R.M. Stress response protein GJA1-20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury. JCI Insight 2018. [Google Scholar] [CrossRef] [Green Version]
- Basheer, W.A.; Xiao, S.; Epifantseva, I.; Fu, Y.; Kleber, A.G.; Hong, T.; Shaw, R.M. GJA1-20k Arranges Actin to Guide Cx43 Delivery to Cardiac Intercalated Discs. Circ. Res. 2017, 121, 1069–1080. [Google Scholar]
- Pfeifer, I.; Anderson, C.; Werner, R.; Oltra, E. Redefining the structure of the mouse connexin43 gene: Selective promoter usage and alternative splicing mechanisms yield transcripts with different translational efficiencies. Nucleic Acids Res. 2004, 32, 4550–4562. [Google Scholar]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; Von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.-H.; Landström, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nature 2008, 10, 1199–1207. [Google Scholar]
- Villarreal, F.J.; Dillmann, W.H. Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am. J. Physiol. Circ. Physiol. 1992, 262, H1861–H1866. [Google Scholar]
- Zhang, D.; Gaussin, V.; Taffet, G.E.; Belaguli, N.S.; Yamada, M.; Schwartz, R.J.; Michael, L.H.; Overbeek, P.; Schneider, M. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 2000, 6, 556–563. [Google Scholar] [PubMed]
- Wang, Y.; Huang, S.; Sah, V.P.; Ross, J.; Brown, J.H.; Han, J.; Chien, K.R. Cardiac Muscle Cell Hypertrophy and Apoptosis Induced by Distinct Members of the p38 Mitogen-activated Protein Kinase Family. J. Biol. Chem. 1998, 273, 2161–2168. [Google Scholar]
- Hsieh, C.-C.; Papaconstantinou, J. The effect of aging on p38 signaling pathway activity in the mouse liver and in response to ROS generated by 3-nitropropionic acid. Mech. Ageing Dev. 2002, 123, 1423–1435. [Google Scholar]
- Hsieh, C.-C.; Rosenblatt, J.I.; Papaconstantinou, J. Age-associated changes in SAPK/JNK and p38 MAPK signaling in response to the generation of ROS by 3-nitropropionic acid. Mech. Ageing Dev. 2003, 124, 733–746. [Google Scholar]
- Simkin, D.; Cavanaugh, E.J.; Kim, D. Control of the single channel conductance of K2P10.1 (TREK-2) by the amino-terminus: Role of alternative translation initiation. J. Physiol. 2008, 586, 5651–5663. [Google Scholar]
- Thomas, D.; Plant, L.D.; Wilkens, C.M.; McCrossan, Z.A.; Goldstein, S.A.N. Alternative Translation Initiation in Rat Brain Yields K2P2.1 Potassium Channels Permeable to Sodium. Neuron 2008, 58, 859–870. [Google Scholar]
- Eckert, M.; Egenberger, B.; Döring, F.; Wischmeyer, E. TREK-1 isoforms generated by alternative translation initiation display different susceptibility to the antidepressant fluoxetine. Neuropharmacology 2011, 61, 918–923. [Google Scholar]
- Kisselbach, J.; Seyler, C.; A Schweizer, P.; Gerstberger, R.; Becker, R.; A Katus, H.; Thomas, D. Modulation of K2P2.1 and K2P10.1 K+channel sensitivity to carvedilol by alternative mRNA translation initiation. Br. J. Pharmacol. 2014, 171, 5182–5194. [Google Scholar]
- Desai, P.N.; Zhang, X.; Wu, S.; Janoshazi, A.; Bolimuntha, S.; Putney, J.; Trebak, M. Multiple types of calcium channels arising from alternative translation initiation of theOrai1message. Sci. Signal. 2015. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J. Cell Sci. 2012, 125, 4354–4361. [Google Scholar] [PubMed] [Green Version]
- Fu, Y.; Zhang, S.-S.; Xiao, S.; Basheer, W.A.; Baum, R.; Epifantseva, I.; Hong, T.; Shaw, R.M. Cx43 Isoform GJA1-20k Promotes Microtubule Dependent Mitochondrial Transport. Front. Physiol. 2017, 8, 905. [Google Scholar] [PubMed]
- Gomez-Ospina, N.; Tsuruta, F.; Barreto-Chang, O.; Hu, L.; Dolmetsch, R. The C Terminus of the L-Type Voltage-Gated Calcium Channel CaV1.2 Encodes a Transcription Factor. Cell 2006, 127, 591–606. [Google Scholar] [PubMed] [Green Version]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [PubMed]
- Sanz, E.; Yang, L.; Su, T.; Morris, D.R.; McKnight, G.S.; Amieux, P.S. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 13939–13944. [Google Scholar] [PubMed] [Green Version]
- Hornstein, N.; Torres, D.; Das Sharma, S.; Tang, G.; Canoll, P.; Sims, P.A. Ligation-free ribosome profiling of cell type-specific translation in the brain. Genome Biol. 2016, 17, 149. [Google Scholar]
- Gao, X.; Wan, J.; Liu, B.; Ma, M.; Shen, B.; Qian, S.-B. Quantitative profiling of initiating ribosomes in vivo. Nat. Methods 2014, 12, 147–153. [Google Scholar]
- Doroudgar, S.; Hofmann, C.; Boileau, E.; Malone, B.; Riechert, E.; Gorska, A.A.; Jakobi, T.; Sandmann, C.; Jürgensen, L.; Kmietczyk, V.; et al. Monitoring Cell-Type-Specific Gene Expression Using Ribosome Profiling In Vivo During Cardiac Hemodynamic Stress. Circ. Res. 2019, 125, 431–448. [Google Scholar]
- Wang, X.; Zhao, B.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar]
- Meyer, K.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar]
- Mathiyalagan, P.; Adamiak, M.; Mayourian, J.; Sassi, Y.; Liang, Y.; Agarwal, N.; Jha, D.; Zhang, S.; Kohlbrenner, E.; Chepurko, E.; et al. FTO-Dependent N6-Methyladenosine Regulates Cardiac Function During Remodeling and Repair. Circulation 2019, 139, 518–532. [Google Scholar] [PubMed]
- Dorn, L.E.; Lasman, L.; Chen, J.; Xu, X.; Hund, T.J.; Medvedovic, M.; Hanna, J.H.; Van Berlo, J.H.; Accornero, F. The N6-Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy. Circulation 2018, 139, 533–545. [Google Scholar]
- Kmietczyk, V.; Riechert, E.; Kalinski, L.; Boileau, E.; Malovrh, E.; Malone, B.; Gorska, A.; Hofmann, C.; Varma, E.; Jürgensen, L.; et al. m6A-mRNA methylation regulates cardiac gene expression and cellular growth. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeitz, M.J.; Smyth, J.W. Translating Translation to Mechanisms of Cardiac Hypertrophy. J. Cardiovasc. Dev. Dis. 2020, 7, 9. https://doi.org/10.3390/jcdd7010009
Zeitz MJ, Smyth JW. Translating Translation to Mechanisms of Cardiac Hypertrophy. Journal of Cardiovascular Development and Disease. 2020; 7(1):9. https://doi.org/10.3390/jcdd7010009
Chicago/Turabian StyleZeitz, Michael J., and James W. Smyth. 2020. "Translating Translation to Mechanisms of Cardiac Hypertrophy" Journal of Cardiovascular Development and Disease 7, no. 1: 9. https://doi.org/10.3390/jcdd7010009
APA StyleZeitz, M. J., & Smyth, J. W. (2020). Translating Translation to Mechanisms of Cardiac Hypertrophy. Journal of Cardiovascular Development and Disease, 7(1), 9. https://doi.org/10.3390/jcdd7010009