
How Cardiac Embryology Translates into Clinical Arrhythmias




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
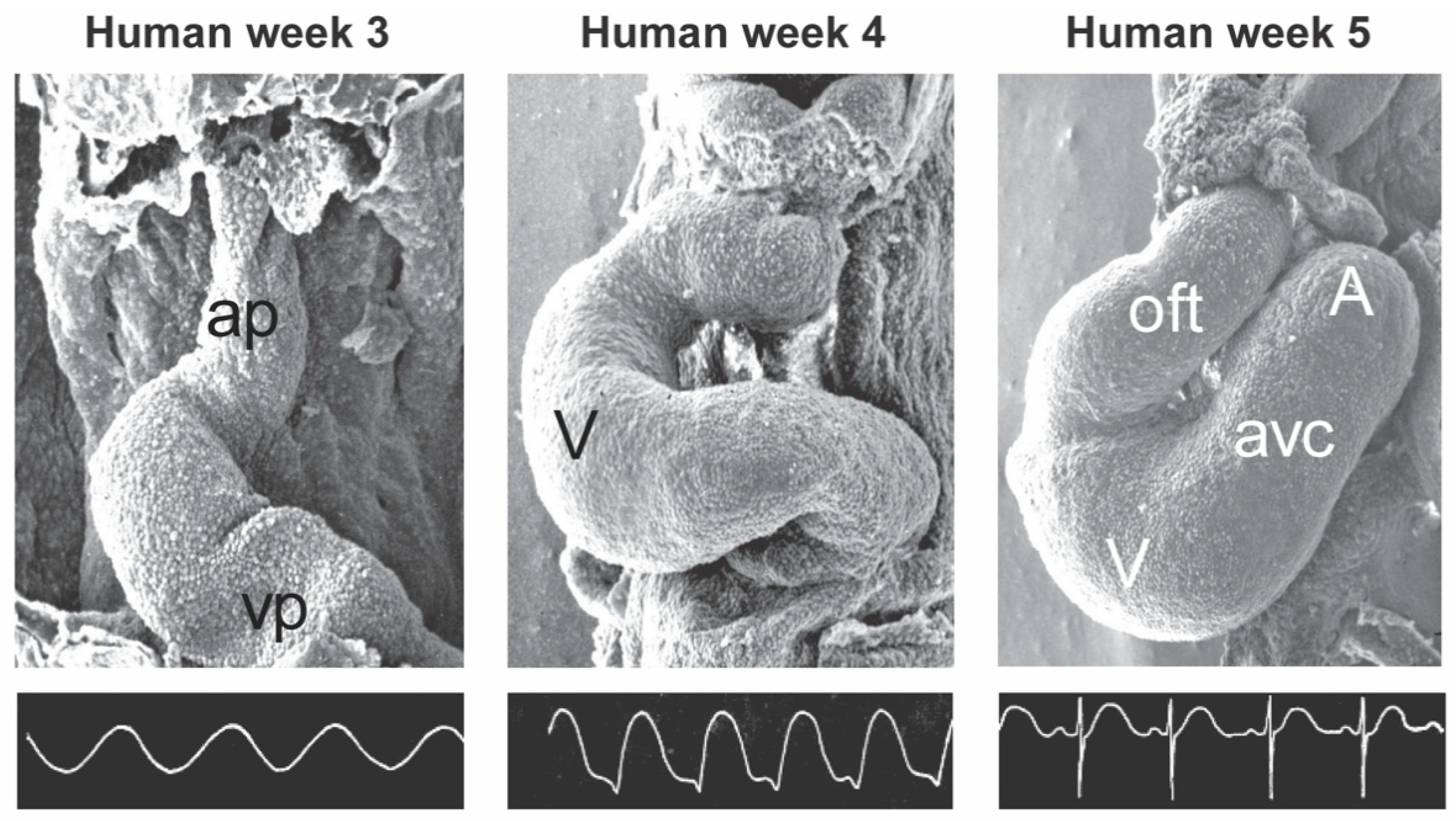
2. General Cardiac Development
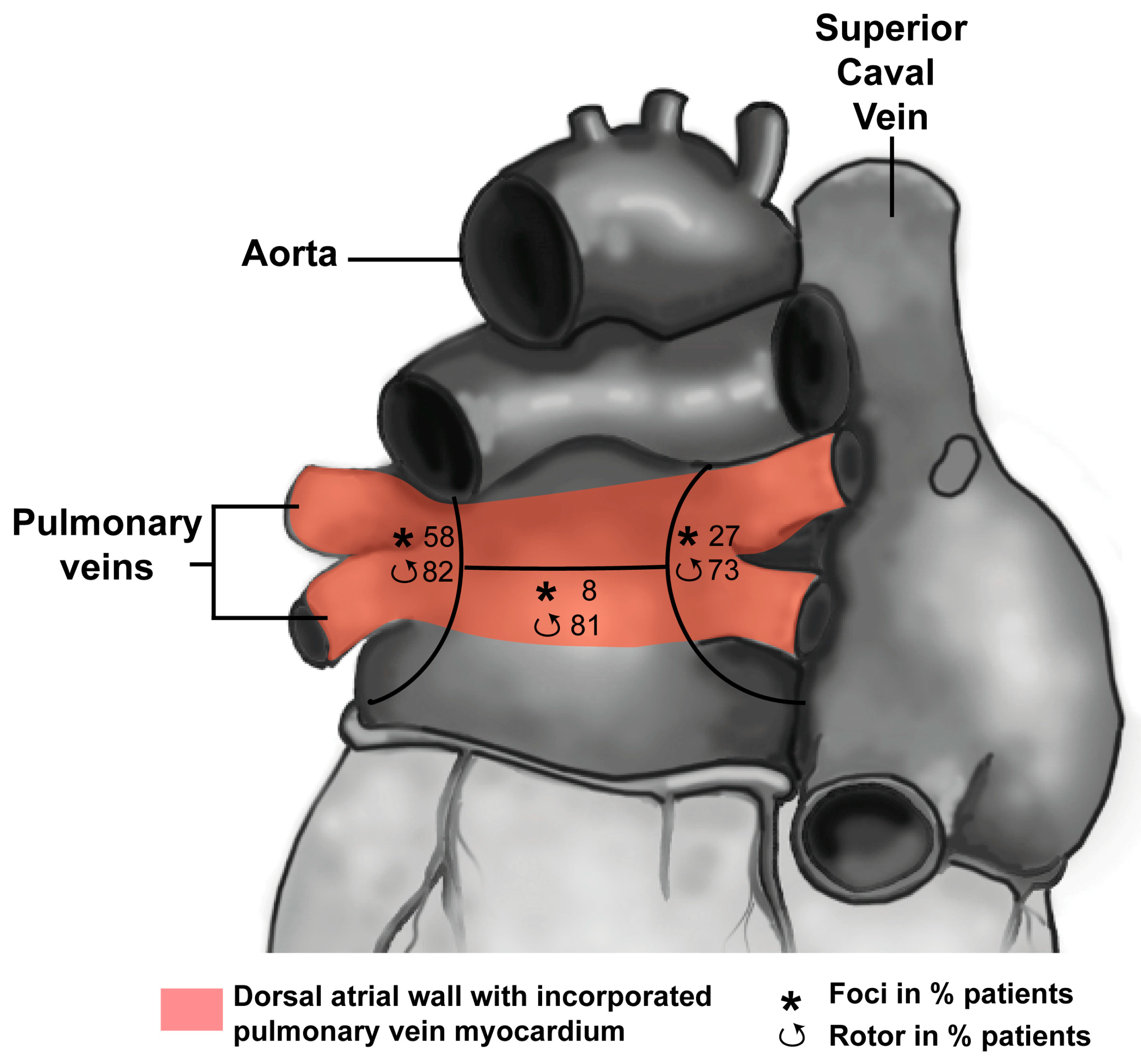
3. Development of the Pulmonary Vein Myocardium and Atrial Arrhythmias
3.1. Development of the Pulmonary Vein Myocardium
3.2. Pulmonary Vein Myocardium and Arrhythmias
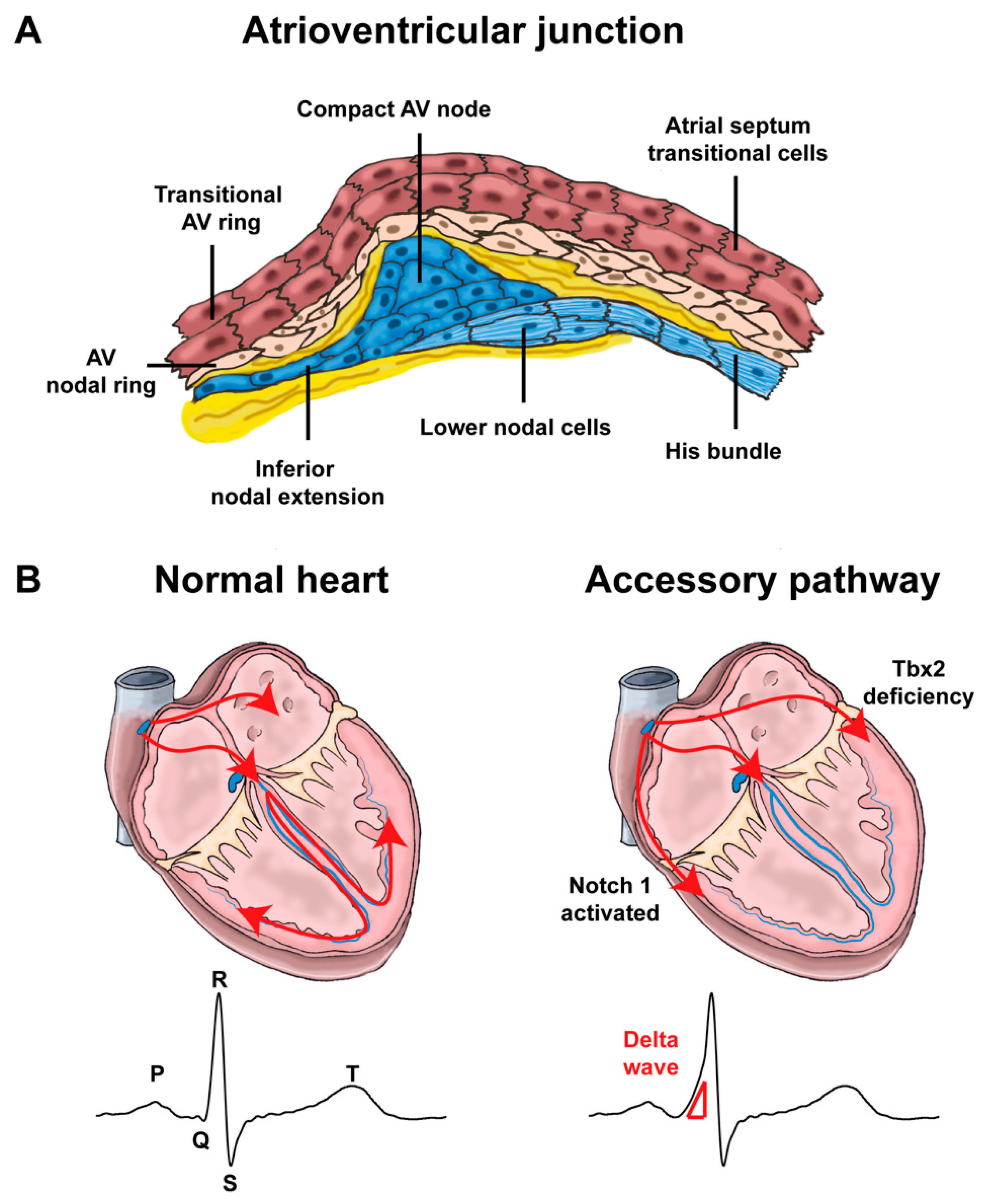
4. Development of Atrioventricular Canal and Accessory Pathways Formation
4.1. Development of the Atrioventricular Canal
4.2. Mispatterning of the Atrioventricular Canal and Arrhythmias
4.2.1. Atrioventricular Conduction Disorders
4.2.2. Re-Entry Tachycardias Involving the Atrioventricular Junction
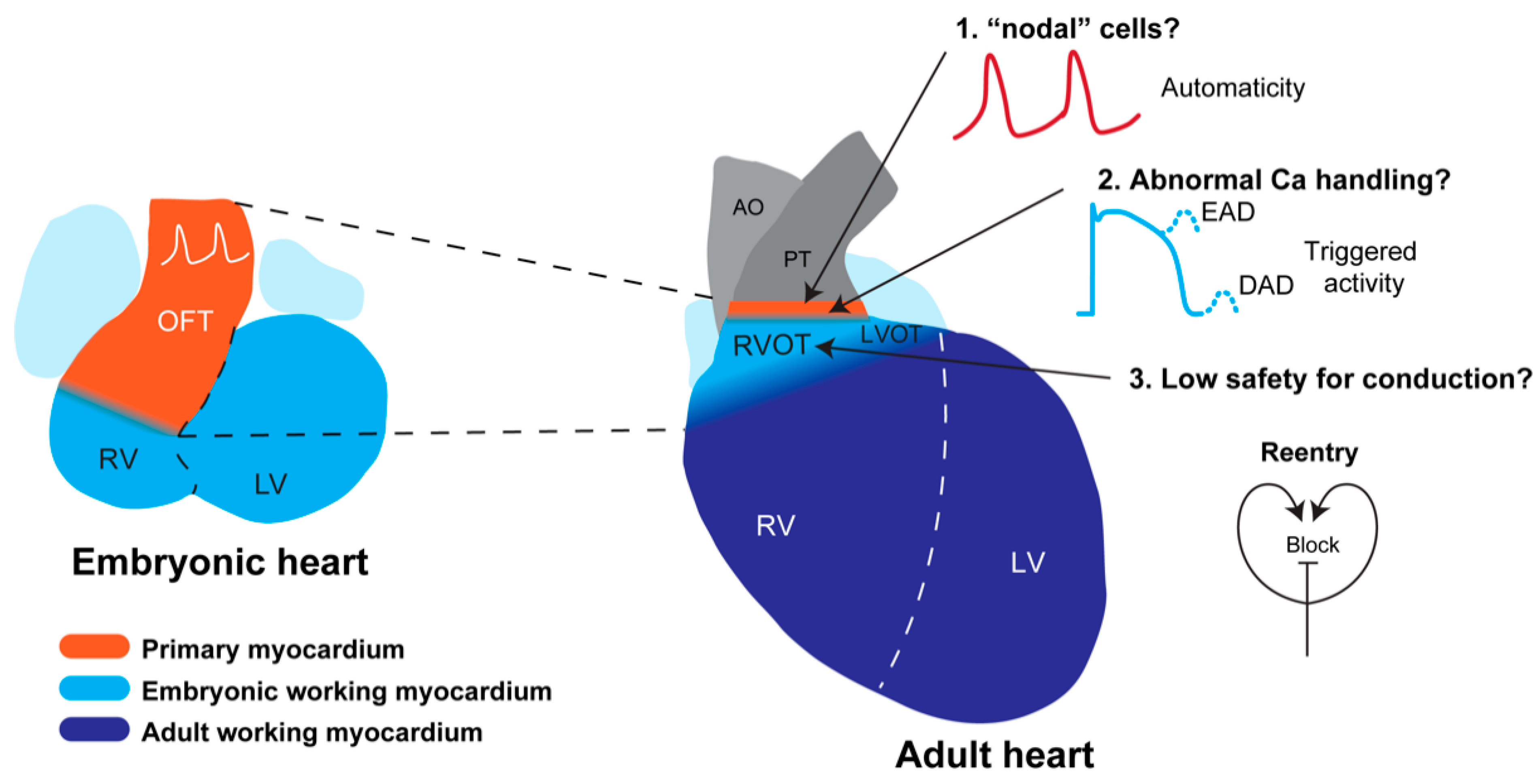
5. Developmental Basis for RVOT Arrhythmias
5.1. Development of the RVOT
5.2. Predisposition of the RVOT for Arrhythmias
5.3. Brugada Syndrome
5.4. Idiopathic Outflow Tract Tachycardia
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Costantini, D.L.; Arruda, E.P.; Agarwal, P.; Kim, K.H.; Zhu, Y.; Zhu, W.; Lebel, M.; Cheng, C.W.; Park, C.Y.; Pierce, S.A.; et al. The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell 2005, 123, 347–358. [Google Scholar] [CrossRef]
- Veerman, C.C.; Podliesna, S.; Tadros, R.; Lodder, E.M.; Mengarelli, I.; de Jonge, B.; Beekman, L.; Barc, J.; Wilders, R.; Wilde, A.A.M.M.; et al. The Brugada Syndrome Susceptibility Gene HEY2 Modulates Cardiac Transmural Ion Channel Patterning and Electrical Heterogeneity. Circ. Res. 2017, 121, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R.; Thakuria, J.V.; Cox, G.F.; Wang, X.; Bi, W.; Bray, M.S.; Shaw, C.; Cheung, S.W.; Chinault, A.C.; Boggs, B.A.; et al. 20p12.3 microdeletion predisposes to Wolff-Parkinson-White syndrome with variable neurocognitive deficits. J. Med. Genet. 2009, 46, 168–175. [Google Scholar] [CrossRef]
- Le, G.L.; Pichon, O.; Isidor, B.; Boceno, M.; Rival, J.M.; David, A.; Le, C.C. A 8.26Mb deletion in 6q16 and a 4.95Mb deletion in 20p12 including JAG1 and BMP2 in a patient with Alagille syndrome and Wolff-Parkinson-White syndrome. Eur.J. Med. Genet. 2008, 51, 651–657. [Google Scholar]
- Basson, C.T.; Huang, T.; Lin, R.C.; Bachinsky, D.R.; Weremowicz, S.; Vaglio, A.; Bruzzone, R.; Quadrelli, R.; Lerone, M.; Romeo, G.; et al. Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc. Natl. Acad. Sci. USA 1999, 96, 2919–2924. [Google Scholar] [CrossRef]
- Hatcher, C.J.; Basson, C.T. Specification of the cardiac conduction system by transcription factors. Circ. Res. 2009, 105, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Schott, J.J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Smits, G.J.; Kispert, A.; Moorman, A.F. Development of the pacemaker tissues of the heart. Circ. Res. 2010, 106, 240–254. [Google Scholar] [CrossRef]
- De Jong, F.; Opthof, T.; Wilde, A.A.; Janse, M.J.; Charles, R.; Lamers, W.H.; Moorman, A.F. Persisting zones of slow impulse conduction in developing chicken hearts. Circ. Res. 1992, 71, 240–250. [Google Scholar] [CrossRef]
- Hoff, E.C.; Kramer, T.C.; DuBois, D.; Patten, B.M. The development of the electrocardiogram of the embryonic heart. Am. Heart J. 1939, 17, 470–488. [Google Scholar] [CrossRef]
- Kelly, R.G.; Brown, N.A.; Buckingham, M.E. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell 2001, 1, 435–440. [Google Scholar] [CrossRef]
- Moorman, A.F.; Christoffels, V.M. Cardiac chamber formation: Development, genes, and evolution. Physiol. Rev. 2003, 83, 1223–1267. [Google Scholar] [CrossRef] [PubMed]
- Boukens, B.J.J.; Rivaud, M.R.R.; Rentschler, S.; Coronel, R. Misinterpretation of the mouse ECG: “musing the waves of Mus musculus. ” J. Physiol. 2014, 592, 4613–4626. [Google Scholar] [CrossRef] [PubMed]
- Sizarov, A.; Ya, J.; de Boer, B.A.; Lamers, W.H.; Christoffels, V.M.; Moorman, A.F. Formation of the building plan of the human heart: Morphogenesis, growth, and differentiation. Circulation 2011, 123, 1125–1135. [Google Scholar] [CrossRef]
- Mommersteeg, M.T.M.; Hoogaars, W.M.H.; Prall, O.W.J.; de Gier-de Vries, C.; Wiese, C.; Clout, D.E.W.; Papaioannou, V.E.; Brown, N.A.; Harvey, R.P.; Moorman, A.F.M.; et al. Molecular pathway for the localized formation of the sinoatrial node. Circ. Res. 2007, 100, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Jongbloed, M.R.M.; Vicente Steijn, R.; Hahurij, N.D.; Kelder, T.P.; Schalij, M.J.; Gittenberger-de Groot, A.C.; Blom, N.A. Normal and abnormal development of the cardiac conduction system; implications for conduction and rhythm disorders in the child and adult. Differentiation 2012, 84, 131–148. [Google Scholar] [CrossRef]
- Lescroart, F.; Mohun, T.; Meilhac, S.M.; Bennett, M.; Buckingham, M. Lineage tree for the venous pole of the heart: Clonal analysis clarifies controversial genealogy based on genetic tracing. Circ. Res. 2012, 111, 1313–1322. [Google Scholar] [CrossRef]
- Mommersteeg, M.T.; Brown, N.A.; Prall, O.W.; de Gier-de, V.C.; Harvey, R.P.; Moorman, A.F.; Christoffels, V.M. Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ. Res. 2007, 101, 902–909. [Google Scholar] [CrossRef]
- Douglas, Y.L.; Jongbloed, M.R.M.; den Hartog, W.C.E.; Bartelings, M.M.; Bogers, A.J.J.C.; Ebels, T.; DeRuiter, M.C.; Gittenberger-de Groot, A.C. Pulmonary vein and atrial wall pathology in human total anomalous pulmonary venous connection. Int. J. Cardiol. 2009, 134, 302–312. [Google Scholar] [CrossRef]
- Webb, S.; Kanani, M.; Anderson, R.H.; Richardson, M.K.; Brown, N.A. Development of the human pulmonary vein and its incorporation in the morphologically left atrium. Cardiol. Young 2001, 11, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Douglas, Y.L.; Jongbloed, M.R.M.; Gittenberger-De Groot, A.C.; Evers, D.; Dion, R.A.E.; Voigt, P.; Bartelings, M.M.; Schalij, M.J.; Ebels, T.; DeRuiter, M.C. Histology of vascular myocardial wall of left atrial body after pulmonary venous incorporation. Am. J. Cardiol. 2006, 97, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Verheule, S.; Wilson, E.E.; Arora, R.; Engle, S.K.; Scott, L.R.; Olgin, J.E. Tissue structure and connexin expression of canine pulmonary veins. Cardiovasc. Res. 2002, 55, 727–738. [Google Scholar] [CrossRef]
- Ehrlich, J.R.; Cha, T.J.; Zhang, L.; Chartier, D.; Melnyk, P.; Hohnloser, S.H.; Nattel, S. Cellular electrophysiology of canine pulmonary vein cardiomyocytes: Action potential and ionic current properties. J. Physiol. 2003, 551, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, P.; Ehrlich, J.R.; Pourrier, M.; Villeneuve, L.; Cha, T.-J.; Nattel, S. Comparison of ion channel distribution and expression in cardiomyocytes of canine pulmonary veins versus left atrium. Cardiovasc. Res. 2005, 65, 104–116. [Google Scholar] [CrossRef]
- Kannel, W.B.; Benjamin, E.J. Status of the epidemiology of atrial fibrillation. Med. Clin. North Am. 2008, 92, 17–40. [Google Scholar] [CrossRef]
- Haissaguerre, M.; Jais, P.; Shah, D.C.; Takahashi, A.; Hocini, M.; Quiniou, G.; Garrigue, S.; Le, M.A.; Le, M.P.; Clementy, J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. New Engl. J. Med. 1998, 339, 659–666. [Google Scholar] [CrossRef]
- Allessie, M.; Ausma, J.; Schotten, U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 2002, 54, 230–246. [Google Scholar] [CrossRef]
- Gottlieb, L.A.; Dekker, L.R.C.; Coronel, R. The Blinding Period Following Ablation Therapy for Atrial Fibrillation: Proarrhythmic and Antiarrhythmic Pathophysiological Mechanisms. JACC Clin. Electrophysiol. 2021, 7, 416–430. [Google Scholar] [CrossRef]
- Perez-Lugones, A.; McMahon, J.T.; Ratliff, N.B.; Saliba, W.I.; Schweikert, R.A.; Marrouche, N.F.; Saad, E.B.; Navia, J.L.; McCarthy, P.M.; Tchou, P.; et al. Evidence of specialized conduction cells in human pulmonary veins of patients with atrial fibrillation. J. Cardiovasc. Electrophysiol. 2003, 14, 803–809. [Google Scholar] [CrossRef]
- Masani, F. Node-like cells in the myocardial layer of the pulmonary vein of rats: An ultrastructural study. J. Anat. 1986, 145, 133–142. [Google Scholar]
- Andalib, A.; Brugada, R.; Nattel, S. Atrial fibrillation: Evidence for genetically determined disease. Curr. Opin. Cardiol. 2008, 23, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Roelens, I.; De, R.L.; Ovaert, C.; Sluysmans, T.; Devriendt, K.; Brunner, H.G.; Vikkula, M. A novel CSX/NKX2-5 mutation causes autosomal-dominant AV block: Are atrial fibrillation and syncopes part of the phenotype? Eur. J. Hum. Genet. 2006, 14, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Gudbjartsson, D.F.; Arnar, D.O.; Helgadottir, A.; Gretarsdottir, S.; Holm, H.; Sigurdsson, A.; Jonasdottir, A.; Baker, A.; Thorleifsson, G.; Kristjansson, K.; et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 2007, 448, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Kahr, P.C.; Morikawa, Y.; Zhang, M.; Rahmani, M.; Heallen, T.R.; Li, L.; Sun, Z.; Olson, E.N.; Amendt, B.A.; et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 2016, 534, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, A.; Daimi, H.; Lozano-Velasco, E.; Dominguez, J.N.; Caballero, R.; Delpon, E.; Tamargo, J.; Cinca, J.; Hove-Madsen, L.; Aranega, A.E.; et al. PITX2 Insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 2011, 4, 269–279. [Google Scholar] [CrossRef]
- Nadadur, R.D.; Broman, M.T.; Boukens, B.; Mazurek, S.R.; Yang, X.; Van Den Boogaard, M.; Bekeny, J.; Gadek, M.; Ward, T.; Zhang, M.; et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef]
- Tucker, N.R.; Dolmatova, E.V.; Lin, H.; Cooper, R.R.; Ye, J.; Hucker, W.J.; Jameson, H.S.; Parsons, V.A.; Weng, L.C.; Mills, R.W.; et al. Diminished PRRX1 Expression Is Associated With Increased Risk of Atrial Fibrillation and Shortening of the Cardiac Action Potential. Circ. Cardiovasc. Genet. 2017, 10, e001902. [Google Scholar] [CrossRef]
- Bossada, F.; Rivaud, M.; Christoffels, V.; Boukens, B.J.D. Noncoding region regulates Prrx1 and predisposes to atrial arrhythmias. Circ. Res. 2021. [Google Scholar] [CrossRef]
- Haissaguerre, M.; Hocini, M.; Denis, A.; Shah, A.J.; Komatsu, Y.; Yamashita, S.; Daly, M.; Amraoui, S.; Zellerhoff, S.; Picat, M.Q.; et al. Driver domains in persistent atrial fibrillation. Circulation 2014, 130, 530–538. [Google Scholar] [CrossRef]
- Aanhaanen, W.T.J.; Boukens, B.J.D.; Sizarov, A.; Wakker, V.; de Gier-de Vries, C.; van Ginneken, A.C.; Moorman, A.F.M.; Coronel, R.; Christoffels, V.M. Defective Tbx2-dependent patterning of the atrioventricular canal myocardium causes accessory pathway formation in mice. J. Clin. Invest. 2011, 121, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Kreuzberg, M.M.; Sohl, G.; Kim, J.S.; Verselis, V.K.; Willecke, K.; Bukauskas, F.F. Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ. Res. 2005, 96, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.V.; McAnally, J.; Bezprozvannaya, S.; Berry, J.M.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Cx30.2 enhancer analysis identifies Gata4 as a novel regulator of atrioventricular delay. Development 2009, 136, 2665–2674. [Google Scholar] [CrossRef]
- Aanhaanen, W.T.; Moorman, A.F.; Christoffels, V.M. Origin and development of the atrioventricular myocardial lineage: Insight into the development of accessory pathways. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 565–577. [Google Scholar] [CrossRef]
- Hoogaars, W.M.; Engel, A.; Brons, J.F.; Verkerk, A.O.; de Lange, F.J.; Wong, L.Y.; Bakker, M.L.; Clout, D.E.; Wakker, V.; Barnett, P.; et al. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 2007, 21, 1098–1112. [Google Scholar] [CrossRef]
- Ma, L.; Lu, M.F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef]
- Gillers, B.S.; Chiplunkar, A.; Aly, H.; Valenta, T.; Basler, K.; Christoffels, V.M.; Efimov, I.R.; Boukens, B.J.; Rentschler, S. Canonical wnt signaling regulates atrioventricular junction programming and electrophysiological properties. Circ. Res. 2015, 116, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Holm, H.; Gudbjartsson, D.F.; Arnar, D.O.; Thorleifsson, G.; Thorgeirsson, G.; Stefansdottir, H.; Gudjonsson, S.A.; Jonasdottir, A.; Mathiesen, E.B.; Njolstad, I.; et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat. Genet. 2010, 42, 117–122. [Google Scholar] [CrossRef]
- Pfeufer, A.; van Noord, C.; Marciante, K.D.; Arking, D.E.; Larson, M.G.; Smith, A.V.; Tarasov, K.V.; Müller, M.; Sotoodehnia, N.; Sinner, M.F.; et al. Genome-wide association study of PR interval. Nat. Genet. 2010, 42, 153–159. [Google Scholar] [CrossRef]
- Sotoodehnia, N.; Isaacs, A.; de Bakker, P.I.W.; Dörr, M.; Newton-Cheh, C.; Nolte, I.M.; van der Harst, P.; Müller, M.; Eijgelsheim, M.; Alonso, A.; et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet. 2010, 42, 1068–1076. [Google Scholar] [CrossRef]
- Lockhart, M.M.; Phelps, A.L.; van den Hoff, M.J.B.; Wessels, A. The Epicardium and the Development of the Atrioventricular Junction in the Murine Heart. J. Dev. Biol. 2014, 2, 1–17. [Google Scholar] [CrossRef]
- Wessels, A.; Markman, M.W.; Vermeulen, J.L.; Anderson, R.H.; Moorman, A.F.; Lamers, W.H. The development of the atrioventricular junction in the human heart. Circ. Res. 1996, 78, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Aanhaanen, W.T.; Mommersteeg, M.T.; Norden, J.; Wakker, V.; de Gier-de, V.C.; Anderson, R.H.; Kispert, A.; Moorman, A.F.; Christoffels, V.M. Developmental origin, growth, and three-dimensional architecture of the atrioventricular conduction axis of the mouse heart. Circ. Res. 2010, 107, 728–736. [Google Scholar] [CrossRef]
- Aanhaanen, W.T.; Brons, J.F.; Dominguez, J.N.; Rana, M.S.; Norden, J.; Airik, R.; Wakker, V.; de Gier-de, V.C.; Brown, N.A.; Kispert, A.; et al. The Tbx2+ primary myocardium of the atrioventricular canal forms the atrioventricular node and the base of the left ventricle. Circ. Res. 2009, 104, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Viragh, S.; Challice, C.E. The development of the conduction system in the mouse embryo heart. Dev. Biol. 1980, 80, 28–45. [Google Scholar] [CrossRef]
- Jay, P.Y.; Harris, B.S.; Maguire, C.T.; Buerger, A.; Wakimoto, H.; Tanaka, M.; Kupershmidt, S.; Roden, D.M.; Schultheiss, T.M.; O’Brien, T.X.; et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J. Clin. Invest. 2004, 113, 1130–1137. [Google Scholar] [CrossRef]
- Moskowitz, I.P.; Pizard, A.; Patel, V.V.; Bruneau, B.G.; Kim, J.B.; Kupershmidt, S.; Roden, D.; Berul, C.I.; Seidman, C.E.; Seidman, J.G. The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development 2004, 131, 4107–4116. [Google Scholar] [CrossRef]
- Rentschler, S.; Harris, B.S.; Kuznekoff, L.; Jain, R.; Manderfield, L.; Lu, M.M.; Morley, G.E.; Patel, V.V.; Epstein, J.A. Notch signaling regulates murine atrioventricular conduction and the formation of accessory pathways. J. Clin. Invest. 2011, 121, 525–533. [Google Scholar] [CrossRef]
- Greener, I.D.; Monfredi, O.; Inada, S.; Chandler, N.J.; Tellez, J.O.; Atkinson, A.; Taube, M.A.; Billeter, R.; Anderson, R.H.; Efimov, I.R.; et al. Molecular architecture of the human specialised atrioventricular conduction axis. J. Mol. Cell. Cardiol. 2011, 50, 642–651. [Google Scholar] [CrossRef]
- McGuire, M.A.; Janse, M.J.; Ross, D.L. “AV nodal” reentry: Part II: AV nodal, AV junctional, or atrionodal reentry? J. Cardiovasc. Electrophysiol. 1993, 4, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Greener, I.D.; Inada, S.; Nikolski, V.P.; Yamamoto, M.; Hancox, J.C.; Zhang, H.; Billeter, R.; Efimov, I.R.; Dobrzynski, H.; et al. Computer three-dimensional reconstruction of the atrioventricular node. Circ. Res. 2008, 102, 975–985. [Google Scholar] [CrossRef]
- Kelder, T.P.; Vicente-Steijn, R.; Harryvan, T.J.; Kosmidis, G.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; Schalij, M.J.; DeRuiter, M.C.; Jongbloed, M.R.M. The sinus venosus myocardium contributes to the atrioventricular canal: Potential role during atrioventricular node development? J. Cell. Mol. Med. 2015, 19, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Mond, H.G.; Proclemer, A. The 11th World Survey of Cardiac Pacing and Implantable Cardioverter-Defibrillators: Calendar Year 2009-A World Society of Arrhythmia’s Project. Pacing Clin. Electrophysiol. 2011, 34, 1013–1027. [Google Scholar] [CrossRef]
- Lee, S.; Wellens, H.J.J.; Josephson, M.E. Paroxysmal atrioventricular block. Heart Rhythm 2009, 6, 1229–1234. [Google Scholar] [CrossRef]
- Alboni, P.; Holz, A.; Brignole, M. Vagally mediated atrioventricular block: Pathophysiology and diagnosis. Heart 2013, 99, 904–908. [Google Scholar] [CrossRef]
- Chambers, J.C.; Zhao, J.; Terracciano, C.M.N.; Bezzina, C.R.; Zhang, W.; Kaba, R.; Navaratnarajah, M.; Lotlikar, A.; Sehmi, J.S.; Kooner, M.K.; et al. Genetic variation in SCN10A influences cardiac conduction. Nat. Genet. 2010, 42, 149–152. [Google Scholar] [CrossRef]
- van Weerd, J.H.; Mohan, R.A.; van Duijvenboden, K.; Hooijkaas, I.B.; Wakker, V.; Boukens, B.J.; Barnett, P.; Christoffels, V.M. Trait-associated noncoding variant regions affect TBX3 regulation and cardiac conduction. Elife 2020, 9. [Google Scholar] [CrossRef]
- Frank, D.U.; Carter, K.L.; Thomas, K.R.; Burr, R.M.; Bakker, M.L.; Coetzee, W.A.; Tristani-Firouzi, M.; Bamshad, M.J.; Christoffels, V.M.; Moon, A.M. Lethal arrhythmias in Tbx3-deficient mice reveal extreme dosage sensitivity of cardiac conduction system function and homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, E154–E163. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.A.; Bosada, F.M.; van Weerd, J.H.; van Duijvenboden, K.; Wang, J.; Mommersteeg, M.T.M.; Hooijkaas, I.B.; Wakker, V.; de Gier-de Vries, C.; Coronel, R.; et al. T-box transcription factor 3 governs a transcriptional program for the function of the mouse atrioventricular conduction system. Proc. Natl. Acad. Sci. USA 2020, 117, 18617–18626. [Google Scholar] [CrossRef] [PubMed]
- Meijler, F.R.; Janse, M.J. Morphology and electrophysiology of the mammalian atrioventricular node. Physiol. Rev. 1988, 68, 608–647. [Google Scholar] [CrossRef] [PubMed]
- Frisch, D.R.; Kwaku, K.F.; Allocco, D.J.; Zimetbaum, P.J. Atrioventricular nodal reentrant tachycardia in two siblings with Wolfram syndrome. J. Cardiovasc. Electrophysiol. 2006, 17, 1029–1031. [Google Scholar] [CrossRef]
- Hayes, J.J.; Sharma, P.P.; Smith, P.N.; Vidaillet, H.J. Familial atrioventricular nodal reentry tachycardia. Pacing Clin. Electrophysiol. 2004, 27, 73–76. [Google Scholar] [CrossRef]
- Anderson, R.H.; Ho, S.Y.; Gillette, P.C.; Becker, A.E. Mahaim, Kent and abnormal atrioventricular conduction. Cardiovasc. Res. 1996, 31, 480–491. [Google Scholar] [CrossRef]
- Deal, B.J.; Keane, J.F.; Gillette, P.C.; Garson, A., Jr. Wolff-Parkinson-White syndrome and supraventricular tachycardia during infancy: Management and follow-up. J. Am. Coll. Cardiol. 1985, 5, 130–135. [Google Scholar] [CrossRef]
- Boukens, B.J.; Janse, M.J. Brief history of arrhythmia in the WPW syndrome—The contribution of George Ralph Mines. J. Physiol. 2013, 591, 4067–4071. [Google Scholar] [CrossRef]
- Jongbloed, M.R.M.; Wijffels, M.C.E.F.; Schalij, M.J.; Blom, N.A.; Poelmann, R.E.; van der Laarse, A.; Mentink, M.M.T.; Wang, Z.; Fishman, G.I.; Gittenberger-de Groot, A.C. Development of the right ventricular inflow tract and moderator band: A possible morphological and functional explanation for Mahaim tachycardia. Circ. Res. 2005, 96, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Stroud, D.M.; Gaussin, V.; Burch, J.B.; Yu, C.; Mishina, Y.; Schneider, M.D.; Fishman, G.I.; Morley, G.E. Abnormal conduction and morphology in the atrioventricular node of mice with atrioventricular canal targeted deletion of Alk3/Bmpr1a receptor. Circulation 2007, 116, 2535–2543. [Google Scholar] [CrossRef]
- Rana, M.S.; Horsten, N.C.A.; Tesink-Taekema, S.; Lamers, W.H.; Moorman, A.F.M.; van den Hoff, M.J.B. Trabeculated right ventricular free wall in the chicken heart forms by ventricularization of the myocardium initially forming the outflow tract. Circ. Res. 2007, 100, 1000–1007. [Google Scholar] [CrossRef]
- De la Cruz, M.V.; Sanchez, G.C.; Arteaga, M.M.; Arguello, C. Experimental study of the development of the truncus and the conus in the chick embryo. J. Anat. 1977, 123, 661–686. [Google Scholar]
- Theveniau-Ruissy, M.; Dandonneau, M.; Mesbah, K.; Ghez, O.; Mattei, M.G.; Miquerol, L.; Kelly, R.G. The del22q11.2 candidate gene Tbx1 controls regional outflow tract identity and coronary artery patterning. Circ. Res. 2008, 103, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.S.; Théveniau-Ruissy, M.; De Bono, C.; Mesbah, K.; Francou, A.; Rammah, M.; Domínguez, J.N.; Roux, M.; Laforest, B.; Anderson, R.H.; et al. Tbx1 coordinates addition of posterior second heart field progenitor cells to the arterial and venous poles of the heart. Circ. Res. 2014, 115, 790–799. [Google Scholar] [CrossRef]
- Sizarov, A.; Anderson, R.H.; Christoffels, V.M.; Moorman, A.F. Three-dimensional and molecular analysis of the venous pole of the developing human heart. Circulation 2010, 122, 798–807. [Google Scholar] [CrossRef]
- Boukens, B.J.; Sylva, M.; De Gier-De Vries, C.; Remme, C.A.; Bezzina, C.R.; Christoffels, V.M.; Coronel, R. Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ. Res. 2013, 113. [Google Scholar] [CrossRef]
- Boukens, B.J.D.; Christoffels, V.M.; Coronel, R.; Moorman, A.F.M. Developmental Basis for Electrophysiological Heterogeneity in the Ventricular and Outflow Tract Myocardium As a Substrate for Life-Threatening Ventricular Arrhythmias. Circ. Res. 2009, 104, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Veerakul, G.; Chandanamattha, P.; Chaothawee, L.; Ariyachaipanich, A.; Jirasirirojanakorn, K.; Likittanasombat, K.; Bhuripanyo, K.; Ngarmukos, T. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011, 123, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Yan, G.X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. J. Arrhythm. 2016, 32, 315–339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, J.; King, J.H.; Huang, C.L.; Fraser, J.A. Measurement and interpretation of electrocardiographic QT intervals in murine hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1553–H1557. [Google Scholar] [CrossRef]
- Ten Sande, J.N.; Coronel, R.; Conrath, C.E.; Driessen, A.H.; de Groot, J.R.; Tan, H.L.; Nademanee, K.; Wilde, A.A.; de Bakker, J.M.; van Dessel, P.F. ST-Segment Elevation and Fractionated Electrograms in Brugada Syndrome Patients Arise from the Same Structurally Abnormal Subepicardial RVOT Area but Have a Different Mechanism. Circ. Arrhythm. Electrophysiol. 2015. [Google Scholar] [CrossRef]
- Kyndt, F.; Probst, V.; Potet, F.; Demolombe, S.; Chevallier, J.C.; Baro, I.; Moisan, J.P.; Boisseau, P.; Schott, J.J.; Escande, D.; et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 2001, 104, 3081–3086. [Google Scholar] [CrossRef]
- Coronel, R.; Casini, S.; Koopmann, T.T.; Wilms-Schopman, F.J.G.; Verkerk, A.O.; de Groot, J.R.; Bhuiyan, Z.; Bezzina, C.R.; Veldkamp, M.W.; Linnenbank, A.C.; et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005, 112, 2769–2777. [Google Scholar] [CrossRef]
- Frustaci, A.; Priori, S.; Pieroni, M.; Chimenti, C.; Napolitano, C.; Rivolta, I.; Sanna, T.; Bellocci, F.; Russo, M.A. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005, 112, 3680–3687. [Google Scholar] [CrossRef]
- Postema, P.G.; van Dessel, P.F.; de Bakker, J.M.; Dekker, L.R.; Linnenbank, A.C.; Hoogendijk, M.G.; Coronel, R.; Tijssen, J.G.; Wilde, A.A.; Tan, H.L. Slow and discontinuous conduction conspire in Brugada syndrome: A right ventricular mapping and stimulation study. Circ. Arrhythm. Electrophysiol. 2008, 1, 379–386. [Google Scholar] [CrossRef]
- Hoogendijk, M.G.; Potse, M.; Vinet, A.; de Bakker, J.M.; Coronel, R. ST segment elevation by current-to-load mismatch: An experimental and computational study. Heart Rhythm 2011, 8, 111–118. [Google Scholar] [CrossRef]
- Hoogendijk, M.G.; Potse, M.; Linnenbank, A.C.; Verkerk, A.O.; Den Ruijter, H.M.; van Amersfoorth, S.C.; Klaver, E.C.; Beekman, L.; Bezzina, C.R.; Postema, P.G.; et al. Mechanism of right precordial ST-segment elevation in structural heart disease: Excitation failure by current-to-load mismatch. Heart Rhythm 2010, 7, 238–248. [Google Scholar] [CrossRef]
- Hoogendijk, M.G.; Opthof, T.; Postema, P.G.; Wilde, A.A.; de Bakker, J.M.; Coronel, R. The Brugada ECG pattern: A marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2010, 3, 283–290. [Google Scholar] [CrossRef]
- Boukens, B.J.; Coronel, R.; Christoffels, V.M.; Engineering, B. Embryonic development of the right ventricular out fl ow tract and arrhythmias. Heart Rhythm 2016, 13, 616–622. [Google Scholar] [CrossRef]
- Blok, M.; Boukens, B.J. Mechanisms of Arrhythmias in the Brugada Syndrome. Int. J. Mol. Sci. 2020, 21, 7051. [Google Scholar] [CrossRef]
- Behr, E.R.; Ben-Haim, Y.; Ackerman, M.J.; Krahn, A.D.; Wilde, A.A.M. Brugada syndrome and reduced right ventricular outflow tract conduction reserve: A final common pathway? Eur. Heart J. 2021, 42, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Raju, H.; de Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.J.; Iwai, S.; Markowitz, S.M.; Shah, B.K.; Stein, K.M.; Lerman, B.B. Clinical and electrophysiological spectrum of idiopathic ventricular outflow tract arrhythmias. J. Am. Coll. Cardiol. 2007, 49, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Lerman, B.B. Mechanism, diagnosis, and treatment of outflow tract tachycardia. Nat. Rev. Cardiol. 2015, 12, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, W. Arrhythmias originating from the right ventricular outflow tract: How to distinguish “malignant” from “benign”? Heart Rhythm 2009, 6, 1507–1511. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Lin, C.; Li, Y.; Liu, T.; Wang, Y. L-type calcium current in right ventricular outflow tract myocytes of rabbit heart. Sci. China Life Sci. 2012, 55, 41–46. [Google Scholar] [CrossRef][Green Version]
- Lu, Y.-Y.; Chung, F.-P.; Chen, Y.-C.; Tsai, C.-F.; Kao, Y.-H.; Chao, T.-F.; Huang, J.-H.; Chen, S.-A.; Chen, Y.-J. Distinctive electrophysiological characteristics of right ventricular out-flow tract cardiomyocytes. J. Cell. Mol. Med. 2014, 18, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Sato, D.; Garfinkel, A.; Qu, Z.; Weiss, J.N. So little source, so much sink: Requirements for afterdepolarizations to propagate in tissue. Biophys. J. 2010, 99, 1408–1415. [Google Scholar] [CrossRef]
- Dobrzynski, H.; Anderson, R.H.; Atkinson, A.; Borbas, Z.; D’Souza, A.; Fraser, J.F.; Inada, S.; Logantha, S.J.R.J.; Monfredi, O.; Morris, G.M.; et al. Structure, function and clinical relevance of the cardiac conduction system, including the atrioventricular ring and outflow tract tissues. Pharmacol. Ther. 2013, 139, 260–288. [Google Scholar] [CrossRef]
- Liu, C.F.; Cheung, J.W.; Thomas, G.; Ip, J.E.; Markowitz, S.M.; Lerman, B.B. Ubiquitous myocardial extensions into the pulmonary artery demonstrated by integrated intracardiac echocardiography and electroanatomic mapping: Changing the paradigm of idiopathic right ventricular outflow tract arrhythmias. Circ. Arrhythm. Electrophysiol. 2014, 7, 691–700. [Google Scholar] [CrossRef]
- Timmermans, C.; Rodriguez, L.M.; Crijns, H.J.; Moorman, A.F.; Wellens, H.J. Idiopathic left bundle-branch block-shaped ventricular tachycardia may originate above the pulmonary valve. Circulation 2003, 108, 1960–1967. [Google Scholar] [CrossRef]
- Zimmermann, M. Sympathovagal balance prior to onset of repetitive monomorphic idiopathic ventricular tachycardia. Pacing Clin. Electrophysiol. 2005, 28, S163–S167. [Google Scholar] [CrossRef]
- Yoshida, A.; Inoue, T.; Ohnishi, Y.; Yokoyama, M. Heart rate variability before spontaneous episodes of ventricular tachycardia originating from right ventricular outflow tract in patients without organic heart disease. Jpn. Circ. J. 1998, 62, 745–749. [Google Scholar] [CrossRef][Green Version]
- Hayashi, H.; Fujiki, A.; Tani, M.; Mizumaki, K.; Shimono, M.; Inoue, H. Role of sympathovagal balance in the initiation of idiopathic ventricular tachycardia originating from right ventricular outflow tract. Pacing Clin. Electrophysiol. 1997, 20, 2371–2377. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, H.; Dong, R.; Zhao, C.; Yu, T.; Yang, L.; Peng, H.; Wu, Y. Increased Local Sympathetic Nerve Activity During Pathogenesis of Ventricular Arrhythmias Originating from the Right Ventricular Outflow Tract. Med. Sci. Monit. 2017, 23, 1090–1098. [Google Scholar] [CrossRef]
- Chang, H.-Y.; Lo, L.-W.; Chen, Y.-R.; Chou, Y.-H.; Lin, W.-L.; Lin, Y.-J.; Yin, W.-H.; Feng, A.-N.; Chen, S.-A. The autonomic neural mechanism of right ventricular outflow tract tachycardia. Auton. Neurosci. 2018, 212, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, S.; Shimizu, W.; Matsuo, K.; Taguchi, A.; Suyama, K.; Kurita, T.; Aihara, N.; Ohe, T.; Shimomura, K. Localization of optimal ablation site of idiopathic ventricular tachycardia from right and left ventricular outflow tract by body surface ECG. Circulation 1998, 98, 1525–1533. [Google Scholar] [CrossRef]
- Lerman, B.B.; Stein, K.; Engelstein, E.D.; Battleman, D.S.; Lippman, N.; Bei, D.; Catanzaro, D. Mechanism of repetitive monomorphic ventricular tachycardia. Circulation 1995, 92, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Cantillon, D.J.; Kim, R.J.; Markowitz, S.M.; Mittal, S.; Stein, K.M.; Shah, B.K.; Yarlagadda, R.K.; Cheung, J.W.; Tan, V.R.; et al. Right and left ventricular outflow tract tachycardias: Evidence for a common electrophysiologic mechanism. J. Cardiovasc. Electrophysiol. 2006, 17, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Hasdemir, C.; Aydin, H.H.; Celik, H.A.; Simsek, E.; Payzin, S.; Kayikcioglu, M.; Aydin, M.; Kultursay, H.; Can, L.H. Transcriptional profiling of septal wall of the right ventricular outflow tract in patients with idiopathic ventricular arrhythmias. Pacing Clin. Electrophysiol. 2010, 33, 159–167. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivaud, M.R.; Blok, M.; Jongbloed, M.R.M.; Boukens, B.J. How Cardiac Embryology Translates into Clinical Arrhythmias. J. Cardiovasc. Dev. Dis. 2021, 8, 70. https://doi.org/10.3390/jcdd8060070
Rivaud MR, Blok M, Jongbloed MRM, Boukens BJ. How Cardiac Embryology Translates into Clinical Arrhythmias. Journal of Cardiovascular Development and Disease. 2021; 8(6):70. https://doi.org/10.3390/jcdd8060070
Chicago/Turabian StyleRivaud, Mathilde R., Michiel Blok, Monique R. M. Jongbloed, and Bastiaan J. Boukens. 2021. "How Cardiac Embryology Translates into Clinical Arrhythmias" Journal of Cardiovascular Development and Disease 8, no. 6: 70. https://doi.org/10.3390/jcdd8060070
APA StyleRivaud, M. R., Blok, M., Jongbloed, M. R. M., & Boukens, B. J. (2021). How Cardiac Embryology Translates into Clinical Arrhythmias. Journal of Cardiovascular Development and Disease, 8(6), 70. https://doi.org/10.3390/jcdd8060070