The Intriguing Relationships of von Willebrand Factor, ADAMTS13 and Cardiac Disease



Abstract
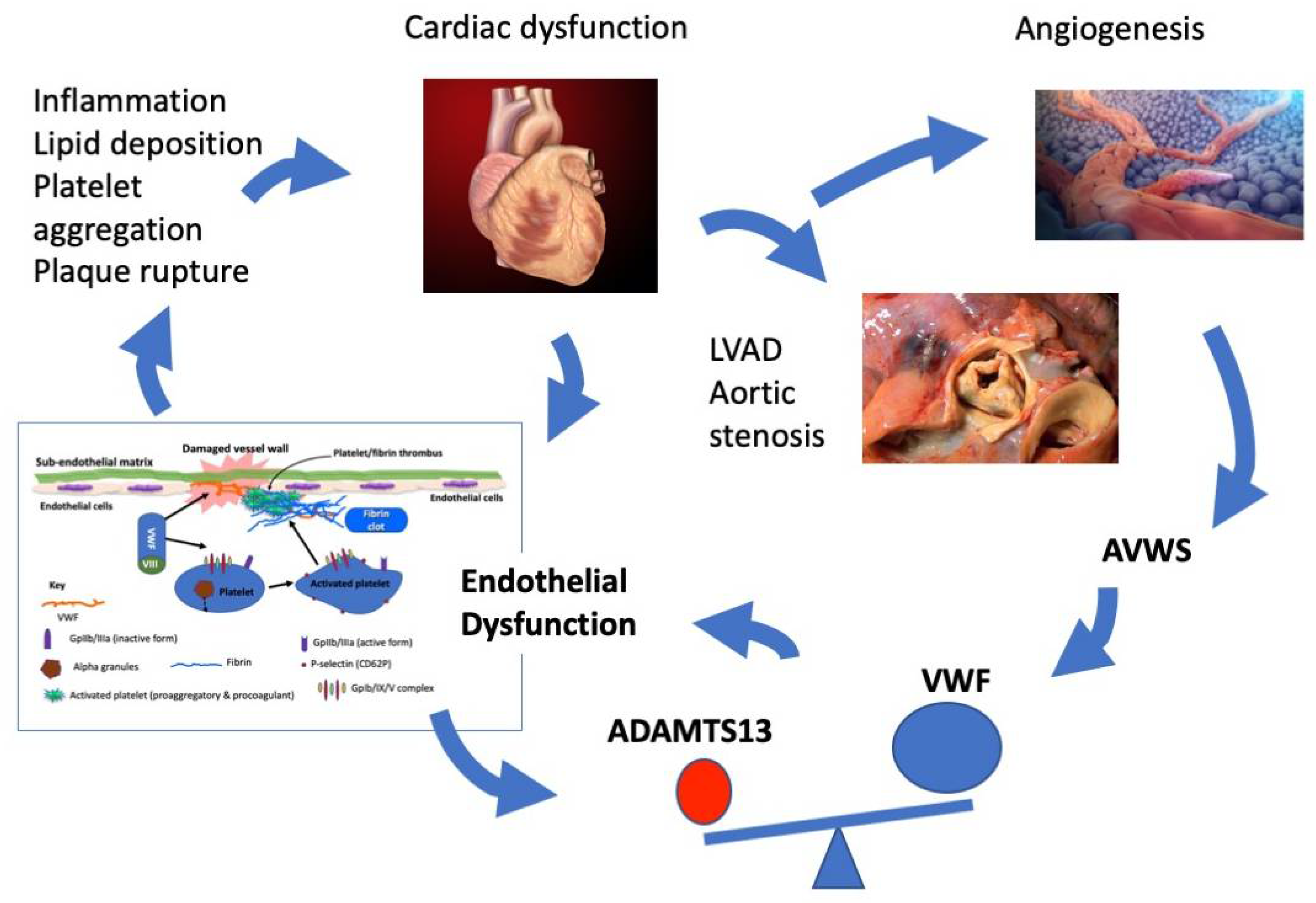
:1. Introduction
2. Endothelial Dysfunction and Cardiac Disease
3. ADAMTS13, VWF and Cardiac Dysfunction
4. Angiogenesis and Acquired von Willebrand Disease
5. Potential Therapeutic Targets
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ho, K.; Pinsky, J.L.; Kannel, W.B.; Levy, D. The epidemiology of heart failure: The Framingham Study. J. Am. Coll. Cardiol. 1993, 22, A6–A13. [Google Scholar] [CrossRef] [Green Version]
- Giannitsi, S.; Bougiakli, M.; Bechlioulis, A.; Naka, K.; Mpougiaklh, M. Endothelial dysfunction and heart failure: A review of the existing bibliography with emphasis on flow mediated dilation. JRSM Cardiovasc. Dis. 2019, 8. [Google Scholar] [CrossRef]
- Reichman-Warmusz, E.; Brzozowa-Zasada, M.; Wojciechowska, C.; Dudek, D.; Warmusz, O.; Wojnicz, R. Decreased immunoreactivity of von Willebrand factor may reflect persistent nature of the endothelial dysfunction in non-ischemic heart failure. Folia Histochem. Cytobiol. 2021, 59, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Salgado, D.R.; Favory, R.; Rocco, J.R.; Silva, E.; Ortiz, J.A.; Donadello, K.; Creteur, J.; Vincent, J.-L.; De Backer, D. Microcirculatory effects of angiotensin II inhibitors in patients with severe heart failure. Clin. Hemorheol. Microcirc. 2013, 54, 87–98. [Google Scholar] [CrossRef]
- Wagner, D. D Cell Biology of von Willebrand Factor. Annu. Rev. Cell Biol. 1990, 6, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Colonne, C.K.; Reardon, B.; Curnow, J.; Favaloro, E.J. Why is Misdiagnosis of von Willebrand Disease Still Prevalent and How Can We Overcome It? A Focus on Clinical Considerations and Recommendations. J. Blood Med. 2021, 12, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, H.; Doman, T.; Kokame, K.; Saiki, Y.; Matsumoto, M. Acquired von Willebrand Syndrome Associated with Cardiovascular Diseases. J. Atheroscler. Thromb. 2019, 26, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Sadler, J.E. Aortic stenosis, von Willebrand factor, and bleeding. N. Engl. J. Med. 2003, 349, 323–325. [Google Scholar] [CrossRef]
- Blackshear, J.L. Heyde Syndrome. Clin. Geriatr. Med. 2019, 35, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. Increased VWF and decreased ADAMTS13 in COVID-19: Creating a milieu for (mi-cro)thrombosis? Semin Thromb Hemost. 2021, 47, 400–418. [Google Scholar]
- Favaloro, E.J. Navigating the Myriad of von Willebrand Factor Assays. Hamostaseologie 2020, 40, 431–442. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Mohammed, S.; Patzke, J. Laboratory Testing for von Willebrand Factor Antigen (VWF:Ag). Methods Mol. Biol. 2017, 1646, 403–416. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Pasalic, L.; Henry, B.; Lippi, G. Laboratory testing for ADAMTS13: Utility for TTP diagnosis/exclusion and beyond. Am. J. Hematol. 2021, 96, 1049–1055. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Mohammed, S.; Chapman, K.; Swanepoel, P.; Zebeljan, D.; Sefhore, O.; Malan, E.; Clifford, J.; Yuen, A.; Donikian, D. A multicentre laboratory assessment of a new automated chemiluminescent assay for ADAMTS13 activity. J. Thromb Haemost. 2021, 19, 417–428. [Google Scholar] [CrossRef]
- Al-Masri, A.A.; Habib, S.S.; Hersi, A.; Al Zamil, H. Effect of Acute Myocardial Infarction on a Disintegrin and Metalloprotease with Thrombospondin Motif 13 and Von Willebrand Factor and Their Relationship with Markers of Inflammation. Int. J. Vasc. Med. 2020, 2020, 4981092. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.; Motto, D.G.; Jensen, M.; Lentz, S.R.; Chauhan, A.K. ADAMTS13 deficiency exacerbates VWF-dependent acute myocardial ischemia/reperfusion injury in mice. Blood 2012, 120, 5224–5230. [Google Scholar] [CrossRef] [Green Version]
- Witsch, T.; Martinod, K.; Sorvillo, N.; Portier, I.; De Meyer, S.F.; Wagner, D.D. Recombinant Human ADAMTS13 Treatment Improves Myocardial Remodeling and Functionality After Pressure Overload Injury in Mice. J. Am. Heart Assoc. 2018, 7, e007004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.; Sörensson, P.; Saleh, N.; Arheden, H.; Rydén, L.; Pernow, J. Circulating endothelial and platelet derived microparticles reflect the size of myocardium at risk in patients with ST-elevation myocardial infarction. Atherosclerosis 2012, 221, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Yang, X.-C.; Jia, X.-W.; Tang, X.-H.; Wang, Z. Von Willebrand factor and ADAMTS13 plasma in older patients with high CHA2DS2-VASc Score with and without atrial fibrillation. Eur. Rev. Med Pharmacol. Sci. 2017, 21, 4907–4912. [Google Scholar]
- Yukhanyan, L.; Freynhofer, M.K.; Siller-Matula, J.M.; Schrör, K.; Huber, K. Genetic variability in response to clopidogrel therapy and its clinical implications. Thromb. Haemost. 2011, 105, S55–S59. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. VWF and ADAMTS13 in COVID-19 and beyond: A question of balance. EMJ Hematol. 2021, 9, 55–68. [Google Scholar]
- Choi, E.J.; Lee, S. A Postoperative Thrombotic Thrombocytopenic Purpura in a Cardiac Surgery Patient: A Case Report. Korean J. Thorac. Cardiovasc. Surg. 2013, 46, 220–222. [Google Scholar] [CrossRef]
- Weinberg, L.; Chang, J.; Hayward, P.; Reynolds, M.; Fernandes, J. Post-Cardiac Surgery Thrombotic Thrombocytopenic Purpura with Digital Ischaemia. Anaesth. Intensiv. Care 2013, 41, 386–389. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.W.; Ting, C.T.; Chen, Y.H.; Wu, T.C.; Hsu, N.W.; Lin, S.J.; Chang, M.S. Differential coronary microvascular function in patients with left ventricular dysfunction of unknown cause—Implication for possible mechanism of myocardial ischemia in early stage of cardiomyopathy. Int. J. Cardiol. 1999, 69, 251–261. [Google Scholar] [CrossRef]
- Hamid, M.A.; Bakhoum, S.W.G.; Sharaf, Y.; Sabry, D.; El-Gengehe, A.T.; Abdel-Latif, A.; Magdy, A.H. Circulating Endothelial Cells and Endothelial Function Predict Major Adverse Cardiac Events and Early Adverse Left Ventricular Remodeling in Patients With ST-Segment Elevation Myocardial Infarction. J. Interv. Cardiol. 2016, 29, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, D.; Rossa, S.; Landmesser, U.; Spiekermann, S.; Engberding, N.; Hornig, B.; Drexler, H. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur. Heart J. 2004, 26, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Neglia, D.; Michelassi, C.; Trivieri, M.G.; Sambuceti, G.; Giorgetti, A.; Pratali, L.; Gallopin, M.; Salvadori, P.; Sorace, O.; Carpeggiani, C.; et al. Prognostic Role of Myocardial Blood Flow Impairment in Idiopathic Left Ventricular Dysfunction. Circulation 2002, 105, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Corban, M.T.; Godo, S.; Burczak, D.R.; Noseworthy, P.A.; Toya, T.; Lewis, B.R.; Lerman, L.O.; Gulati, R.; Lerman, A. Coronary Endothelial Dysfunction Is Associated With Increased Risk of Incident Atrial Fibrillation. J. Am. Heart Assoc. 2020, 9, e014850. [Google Scholar] [CrossRef] [PubMed]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2019, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Hryniewicz, K.; Hriljac, I.; Balidemaj, K.; Dimayuga, C.; Hudaihed, A.; Yasskiy, A. Vascular Endothelial Dysfunction and Mortality Risk in Patients With Chronic Heart Failure. Circulation 2005, 111, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Valentijn, K.M.; Eikenboom, J. Weibel-Palade bodies: A window to von Willebrand disease. J. Thromb. Haemost. 2013, 11, 581–592. [Google Scholar] [CrossRef]
- de Jong, A.; Eikenboom, J.C.J. Developments in the diagnostic procedures for von Willebrand disease. J. Thromb. Haemost. 2016, 14, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gombos, T.; Makó, V.; Cervenak, L.; Papassotiriou, J.; Kunde, J.; Hársfalvi, J.; Förhécz, Z.; Pozsonyi, Z.; Borgulya, G.; Jánoskuti, L.; et al. Levels of von Willebrand factor antigen and von Willebrand factor cleaving protease (ADAMTS13) activity predict clinical events in chronic heart failure. Thromb. Haemost. 2009, 102, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Lip, G.Y.H.; Pearce, L.; Chin, B.S.P.; Conway, D.S.G.; Hart, R.G. Effects of congestive heart failure on plasma von Willebrand factor and soluble P-selectin concentrations in patients with non-valvar atrial fibrillation. Heart 2005, 91, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.R.; Blann, A.D.; Watson, R.D.; Lip, G.Y.H. Abnormalities of hemorheological, endothelial, and platelet function in patients with chronic heart failure in sinus rhythm: Effects of angiotensin converting enzyme inhibitor and beta-blocker therapy. Circulation 2001, 103, 1746–1751. [Google Scholar] [CrossRef] [Green Version]
- Kleber, M.E.; Koller, L.; Goliasch, G.; Sulzgruber, P.; Scharnagl, H.; Silbernagel, G.; Grammer, T.B.; Delgado, G.; Tomaschitz, A.; Pilz, S.; et al. Von Willebrand Factor Improves Risk Prediction in Addition to N-Terminal Pro–B-type Natriuretic Peptide in Patients Referred to Coronary Angiography and Signs and Symptoms of Heart Failure and Preserved Ejection Fraction. Circ. Heart Fail. 2015, 8, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Galen, K.P.M.; Tuinenburg, A.; Smeets, E.M.; Schutgens, R.E.G. Von Willebrand factor deficiency and atherosclerosis. Blood Rev. 2012, 26, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, W.; Hassan, S.; Dabak, V.; Kuriakose, P. Thrombosis in VonWillebrand disease. Thromb. Res. 2012, 130, e255–e258. [Google Scholar] [CrossRef] [PubMed]
- Seaman, C.D.; Yabes, J.; Comer, D.M.; Ragni, M.V. Does deficiency of von Willebrand factor protect against cardiovascular disease? Analysis of a national discharge register. J. Thromb. Haemost. 2015, 13, 1999–2003. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, M.H.; Seaman, C.D.; Comer, D.M.; Yabes, J.G.; Ragni, M.V. Prevalence and Risk Factors Associated With Hypertension in von Willebrand Disease. Clin. Appl. Thromb. 2016, 24, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Saltzman, D.J.; Chang, J.C.; Jimenez, J.C.; Carson, J.G.; Abolhoda, A.; Newman, R.S.; Milliken, J.C. Postoperative Thrombotic Thrombocytopenic Purpura After Open Heart Operations. Ann. Thorac. Surg. 2010, 89, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.B. Thrombotic Thrombocytopenic Purpura and Heparin-Induced Thrombocytopenia: Two Unique Causes of Life-Threatening Thrombocytopenia. Clin. Lab. Med. 2009, 29, 321–338. [Google Scholar] [CrossRef]
- Le Besnerais, M.; Favre, J.; Denis, C.; Mulder, P.; Martinet, J.; Nicol, L.; Levesque, H.; Veyradier, A.; Kopić, A.; Motto, D.G.; et al. Assessment of endothelial damage and cardiac injury in a mouse model mimicking thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2016, 14, 1917–1930. [Google Scholar] [CrossRef] [Green Version]
- Friberg, L.; Rosenqvist, M.; Lip, G.Y. Evaluation of risk stratification schemes for ischaemic stroke and bleeding in 182 678 patients with atrial fibrillation: The Swedish Atrial Fibrillation cohort study. Eur. Heart J. 2012, 33, 1500–1510. [Google Scholar] [CrossRef]
- Singer, D.E.; Albers, G.W.; Dalen, J.E.; Fang, M.C.; Go, A.S.; Halperin, J.L.; Lip, G.; Manning, W.J. Antithrombotic therapy in atrial fi-brillation: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008, 133, 546S–592S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneveld, M.A.; de Maat, M.P.; Leebeek, F.W. Von Willebrand factor and ADAMTS13 in arterial thrombosis: A systematic review and meta-analysis. Blood Rev. 2014, 28, 167–178. [Google Scholar] [CrossRef]
- Newnham, M.; South, K.; Bleda, M.; Auger, W.R.; Barberà, J.A.; Bogaard, H.; Bunclark, K.; Cannon, J.E.; Delcroix, M.; Hadinnapola, C.; et al. The ADAMTS13–VWF axis is dysregulated in chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801805. [Google Scholar] [CrossRef]
- Starke, R.D.; Ferraro, F.; Paschalaki, K.; Dryden, N.H.; McKinnon, T.A.J.; Sutton, R.E.; Payne, E.M.; Haskard, D.O.; Hughes, A.; Cutler, D.; et al. Endothelial von Willebrand factor regulates angiogenesis. Blood 2011, 117, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Stockschlaeder, M.; Schneppenheim, R.; Budde, U. Update on von Willebrand factor multimers: Focus on high-molecular-weight multimers and their role in hemostasis. Blood Coagul. Fibrinolysis 2014, 25, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abshire, T.C.; Federici, A.B.; Alvárez, M.T.; Bowen, J.; Carcao, M.D.; Gill, J.C.; Key, N.S.; Kouides, P.A.; Kurnik, K.; Lail, A.E.; et al. Prophylaxis in severe forms of von Willebrand’s disease: Results from the von Willebrand Disease Prophylaxis Network (VWD PN). Haemophilia 2012, 19, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.F.; Moake, J.L.; Bernardo, A.; Fujikawa, K.; Ball, C.; Nolasco, L.; López, J.A.; Cruz, M.A. ADAMTS-13 Metalloprotease Interacts with the Endothelial Cell-derived Ultra-large von Willebrand Factor. J. Biol. Chem. 2003, 278, 29633–29639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, S.; Kasatkar, P.; Ghosh, K. Pathophysiology of acquired von Willebrand disease: A concise review. Eur. J. Haematol. 2011, 87, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Massyn, M.W.; Khan, S.A. Heyde syndrome: A common diagnosis in older patients with severe aortic stenosis. Age Ageing 2008, 38, 267–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyersdorf, F.; Nakamura, L.; Benk, C.; Berchtold-Herz, M.; Trummer, G.; Schlensak, C.; Heilmann, C.; Geisen, U.; Zieger, B. Acquired von Willebrand syndrome in patients with ventricular assist device or total artificial heart. Thromb. Haemost. 2010, 103, 962–967. [Google Scholar] [CrossRef]
- Schlagenhauf, A.; Kalbhenn, J.; Geisen, U.; Beyersdorf, F.; Zieger, B. Acquired von Willebrand Syndrome and Platelet Function Defects during Extracorporeal Life Support (Mechanical Circulatory Support). Hamostaseologie 2020, 40, 221–225. [Google Scholar] [CrossRef]
- Vincentelli, A.; Susen, S.; Tourneau, T.L.; Six, I.; Fabre, O.; Juthier, F.; Bauters, A.; Decoene, C.; Goudemand, J.; Prat, A.; et al. Acquired von Willebrand Syndrome in Aortic Stenosis. N. Engl. J. Med. 2003, 349, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Hollander, K.N.; Ibekwe, S.O.; Williams, B.; Tanaka, K. Heyde Syndrome–Pathophysiology and Perioperative Implications. J. Cardiothorac. Vasc. Anesthesia 2020, S1053-0770, 31098–31103. [Google Scholar] [CrossRef]
- Wan, S.H.; Liang, J.J.; Vaidya, R.; Blackshear, J.L.; Chen, D. Acquired Von Willebrand Syndrome Secondary to Mitral and Aortic Regurgitation. Can. J. Cardiol. 2014, 30, 1108.e9–1108.e10. [Google Scholar] [CrossRef]
- Mohri, H. Acquired von Willebrand syndrome: Features and management. Am. J. Hematol. 2006, 81, 616–623. [Google Scholar] [CrossRef]
- Ferraro, F.; Patella, F.; Costa, J.R.; Ketteler, R.; Kriston-Vizi, J.; Cutler, D.F. Modulation of endothelial organelle size as an antithrombotic strategy. J. Thromb. Haemost. 2020, 18, 3296–3308. [Google Scholar] [CrossRef] [PubMed]
- Lillicrap, D. Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J. Thromb. Haemost. 2020, 18, 786–787. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Gilbert, J.C.; Hatala, P.; Harvey, W.; Liang, Z.; Gao, S.; Kang, D.; Jilma, B. The development and characterization of a long acting anti-thrombotic von Willebrand factor (VWF) aptamer. J. Thromb. Haemost. 2020, 18, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, K.D.; Buchtele, N.; Schoergenhofer, C.; Derhaschnig, U.; Gelbenegger, G.; Brostjan, C.; Zhu, S.; Gilbert, J.C.; Jilma, B. The aptamer BT200 effectively inhibits von Willebrand factor (VWF) dependent platelet function after stimulated VWF release by desmopressin or endotoxin. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Langhauser, F.; Desender, L.; Vandenbulcke, A.; Rottensteiner, H.; Plaimauer, B.; François, O.; Andersson, T.; Deckmyn, H.; Scheiflinger, F.; et al. ADAMTS13-mediated thrombolysis of t-PA–resistant occlusions in ischemic stroke in mice. Blood 2016, 127, 2337–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Lymphoproliferative Disorders | Monoclonal Gammopathy of Undetermined Significance |
| Multiple Myeloma | |
| Non-Hodgkin’s Lymphoma | |
| Waldenstrom’s Macroglobulinemia | |
| Hairy Cell Leukemia | |
| Myeloproliferative Disorders | Essential Thrombocythemia |
| Polycythemia Vera | |
| Chronic Myeloid Leukemia | |
| Tumours | Wilms’ Tumor |
| Ewing’s Sarcoma | |
| Cardiac Disorders | Aortic Stenosis |
| Left Ventricular Assist Devices | |
| Heart Transplantation | |
| Coronary Artery bypass Surgery | |
| Paravalvular Leak | |
| Hypertrophic Obstructive Cardiomyopathy | |
| Congenital Heart Disease | |
| Autoimmune | SYSTEMATIC Lupus Erythematosus |
| Other Autoimmune Disorders | |
| Drug Induced | Cefotaxime |
| Levofloxacin | |
| Ciprofloxacin | |
| Valproic Acid | |
| Hydroxy Ethyl Starch | |
| High-dose Recombinant Factor VIII | |
| Miscellaneous | Gaucher’s Disease |
| Renal Transplantation | |
| Hypothyroidism | |
| Extracorporeal Membrane Oxygenation Devices |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reardon, B.; Pasalic, L.; Favaloro, E.J. The Intriguing Relationships of von Willebrand Factor, ADAMTS13 and Cardiac Disease. J. Cardiovasc. Dev. Dis. 2021, 8, 115. https://doi.org/10.3390/jcdd8090115
Reardon B, Pasalic L, Favaloro EJ. The Intriguing Relationships of von Willebrand Factor, ADAMTS13 and Cardiac Disease. Journal of Cardiovascular Development and Disease. 2021; 8(9):115. https://doi.org/10.3390/jcdd8090115
Chicago/Turabian StyleReardon, Benjamin, Leonardo Pasalic, and Emmanuel J. Favaloro. 2021. "The Intriguing Relationships of von Willebrand Factor, ADAMTS13 and Cardiac Disease" Journal of Cardiovascular Development and Disease 8, no. 9: 115. https://doi.org/10.3390/jcdd8090115
APA StyleReardon, B., Pasalic, L., & Favaloro, E. J. (2021). The Intriguing Relationships of von Willebrand Factor, ADAMTS13 and Cardiac Disease. Journal of Cardiovascular Development and Disease, 8(9), 115. https://doi.org/10.3390/jcdd8090115