
DCHS1, Lix1L, and the Septin Cytoskeleton: Molecular and Developmental Etiology of Mitral Valve Prolapse

 , , , ,
, , , ,  ,
,  , , and
, , and 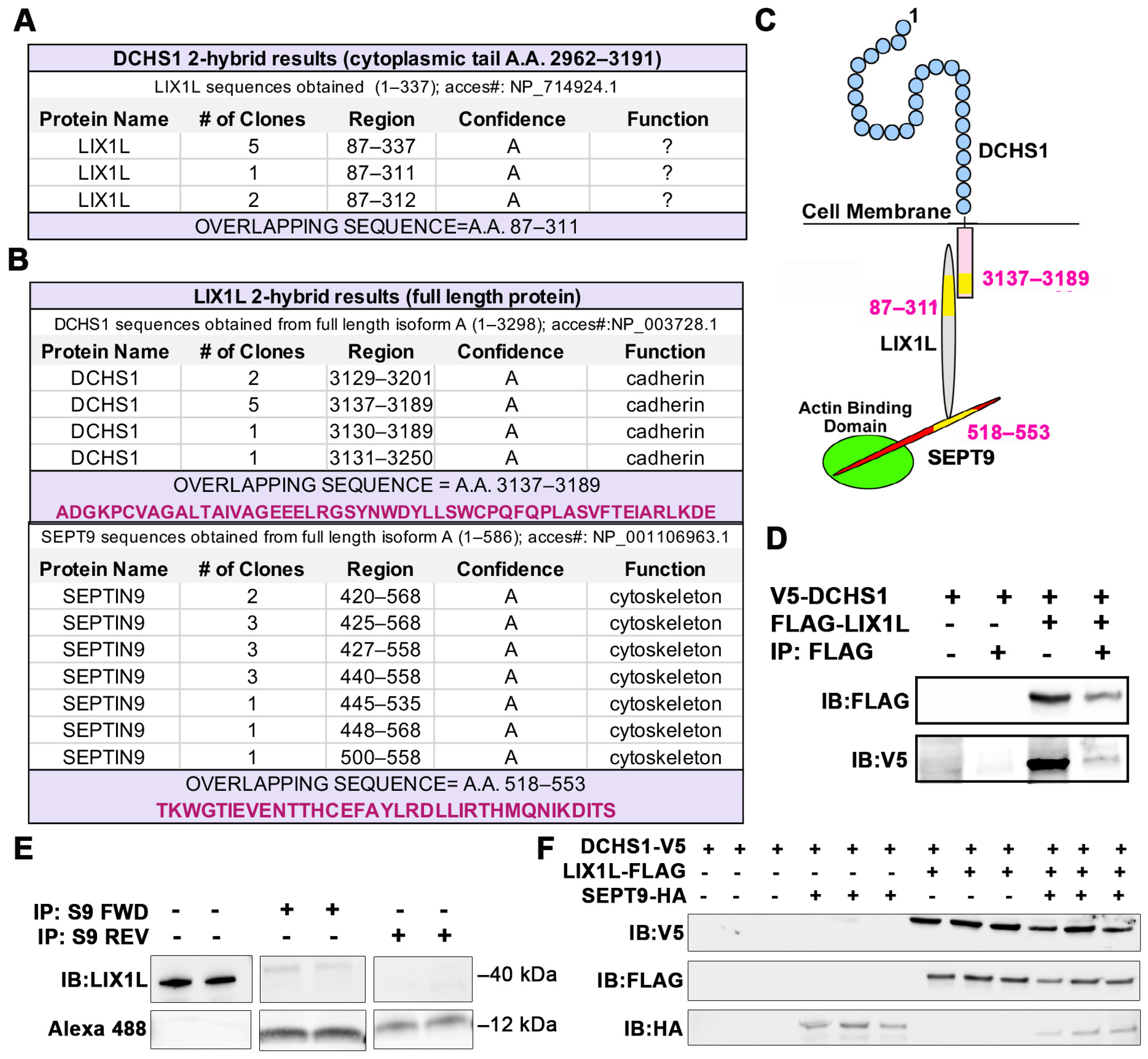{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
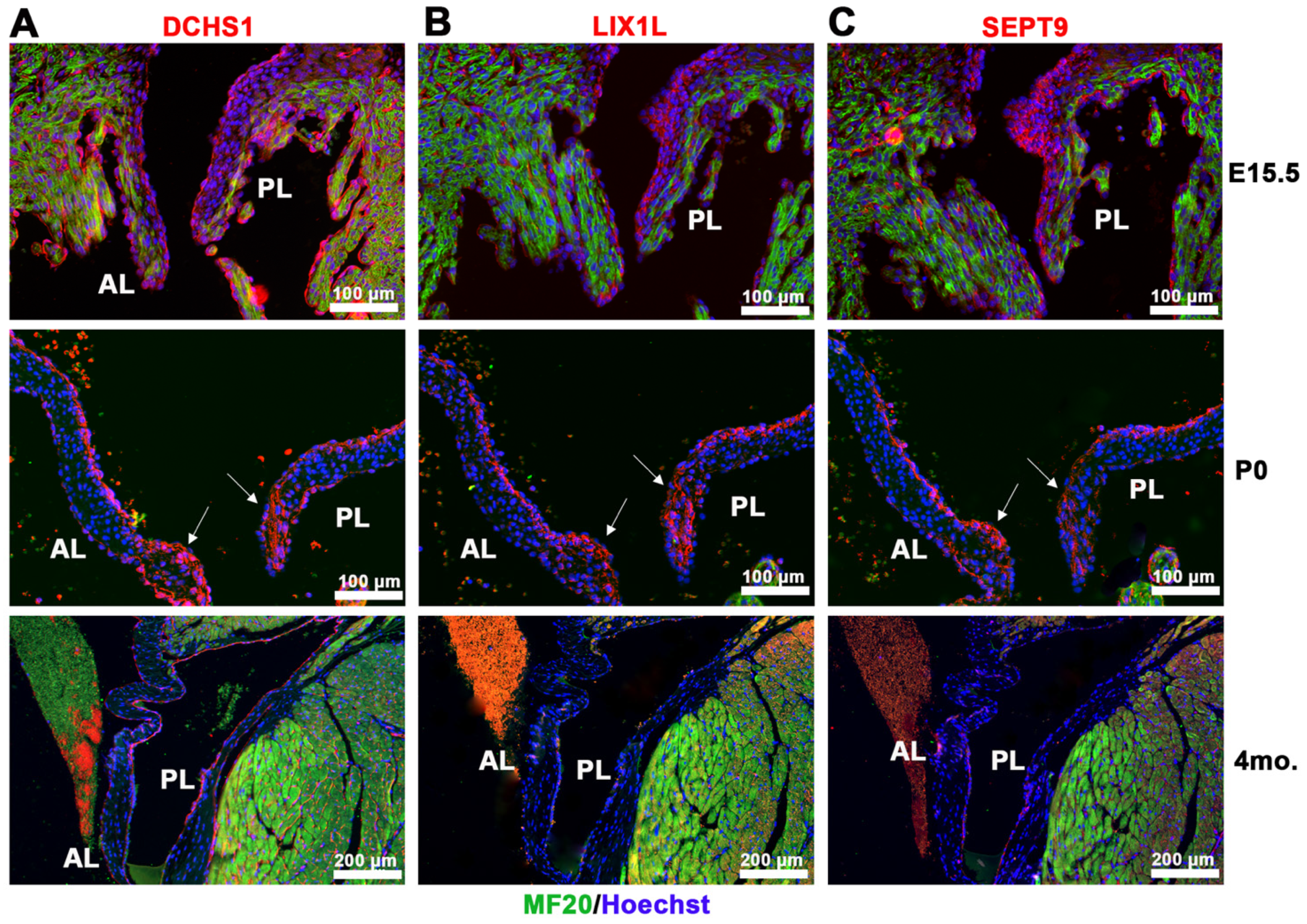
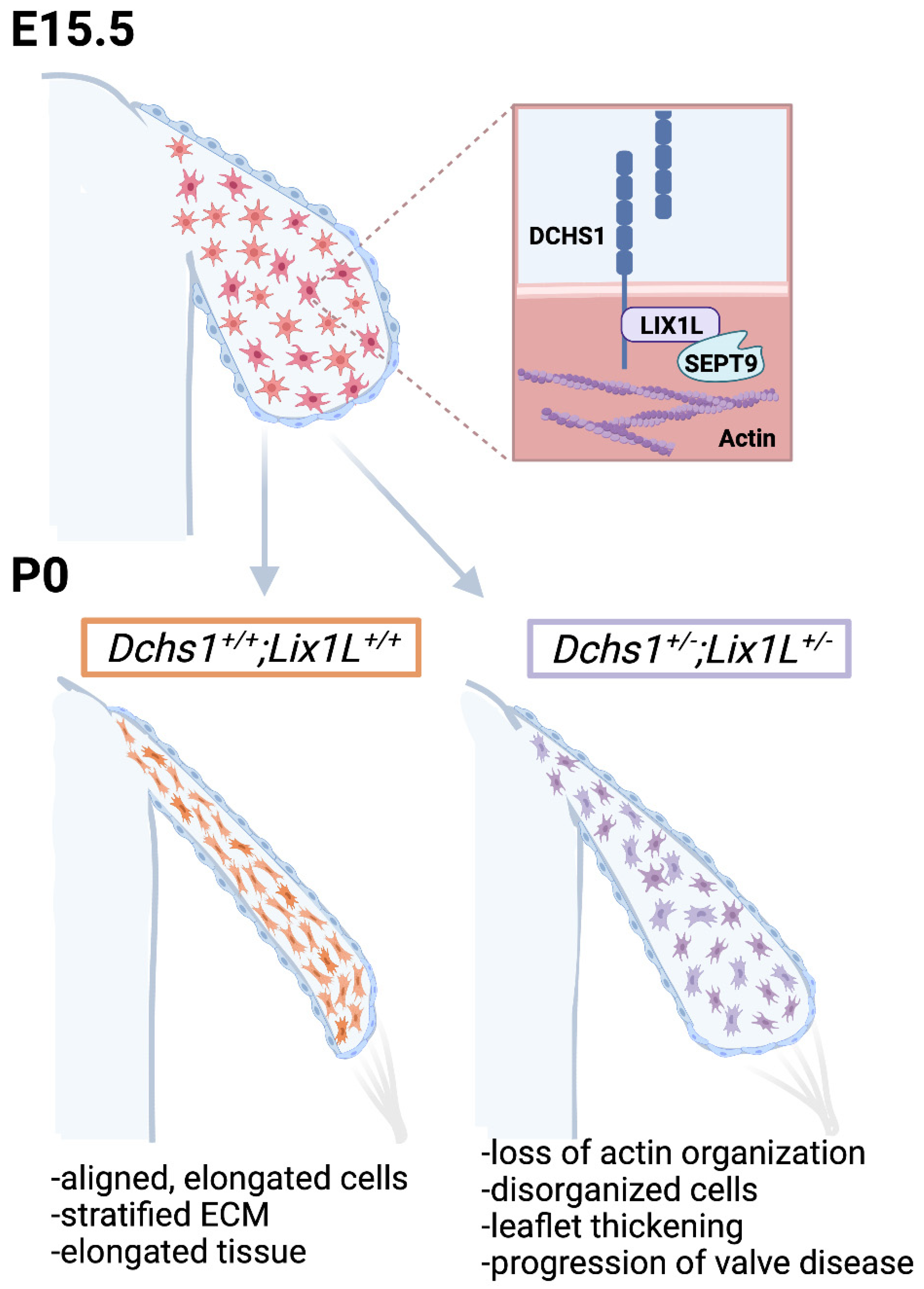
2.1. Identification of DCHS1-LIX1L-SEPT9 (DLS) Protein Complex
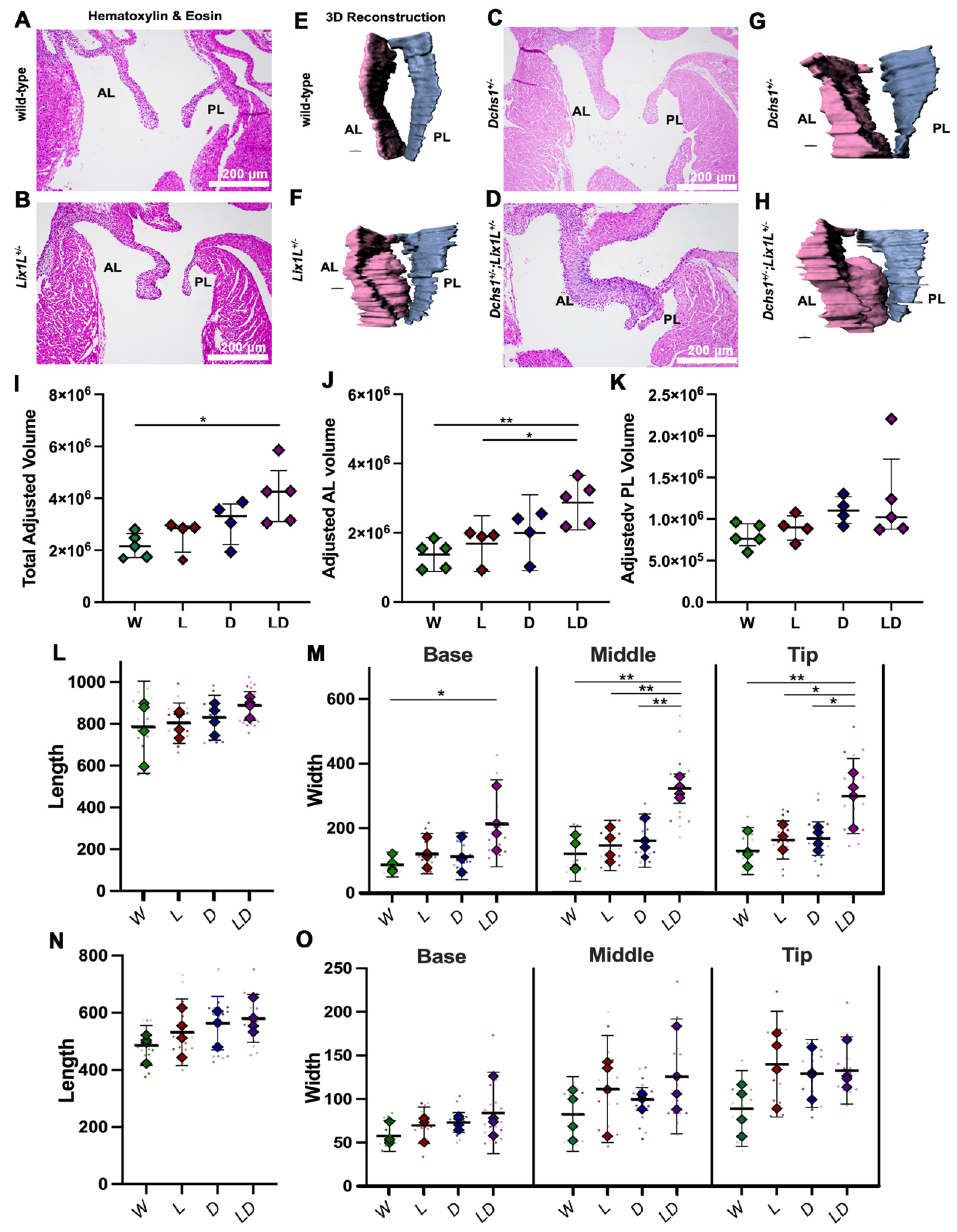
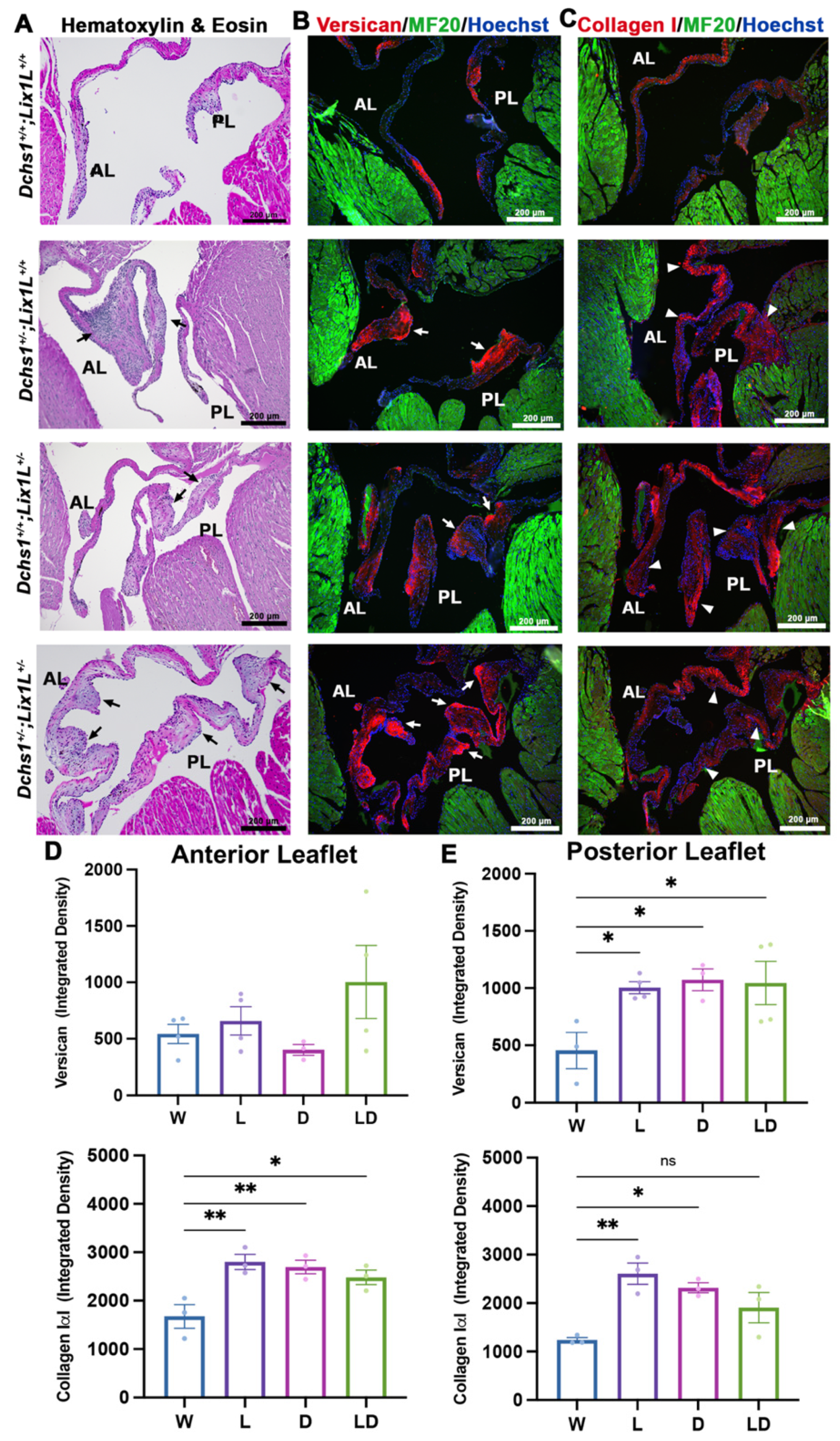
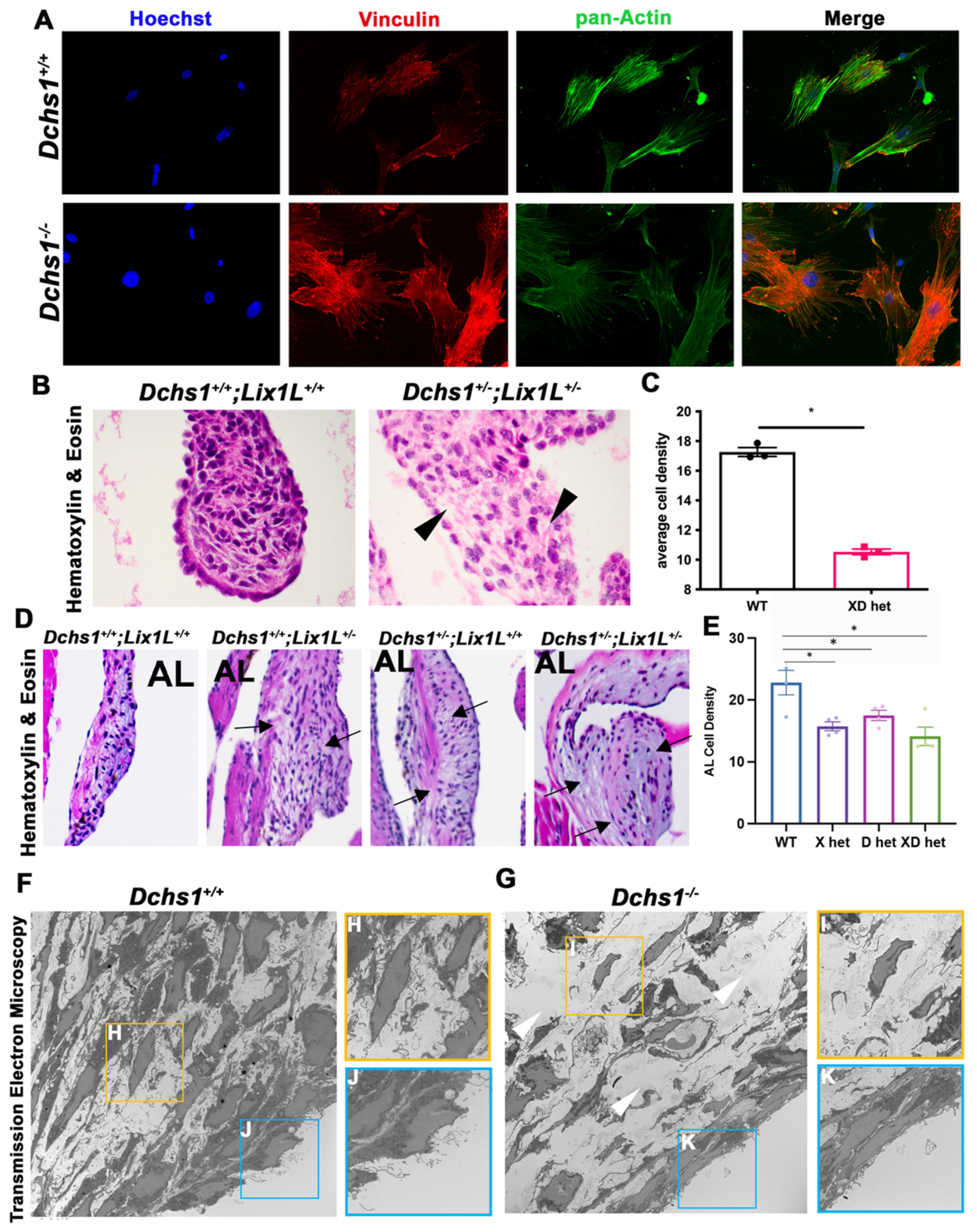
2.2. DCHS1 and Lix1L Genetically Interact during Valve Development
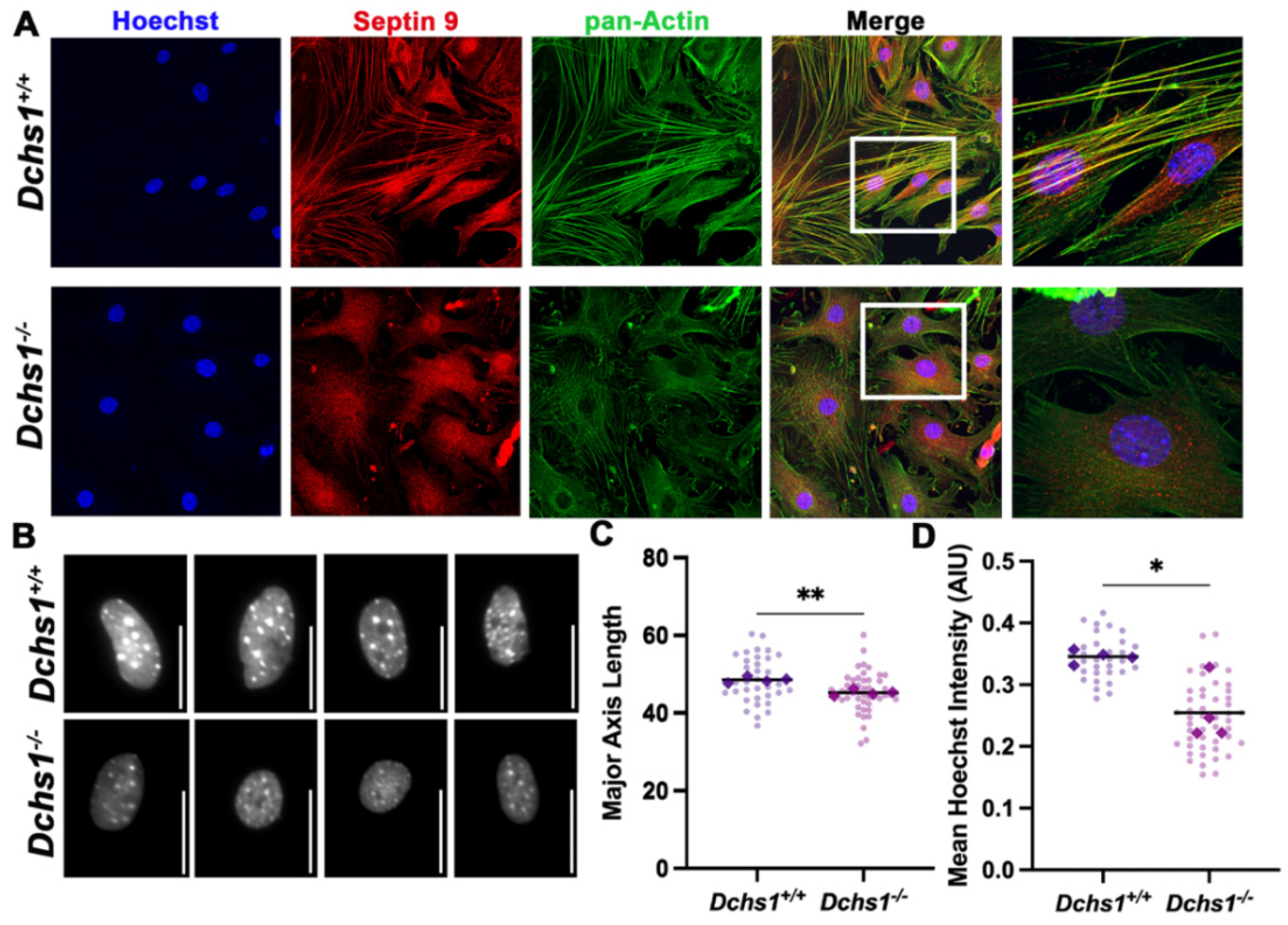
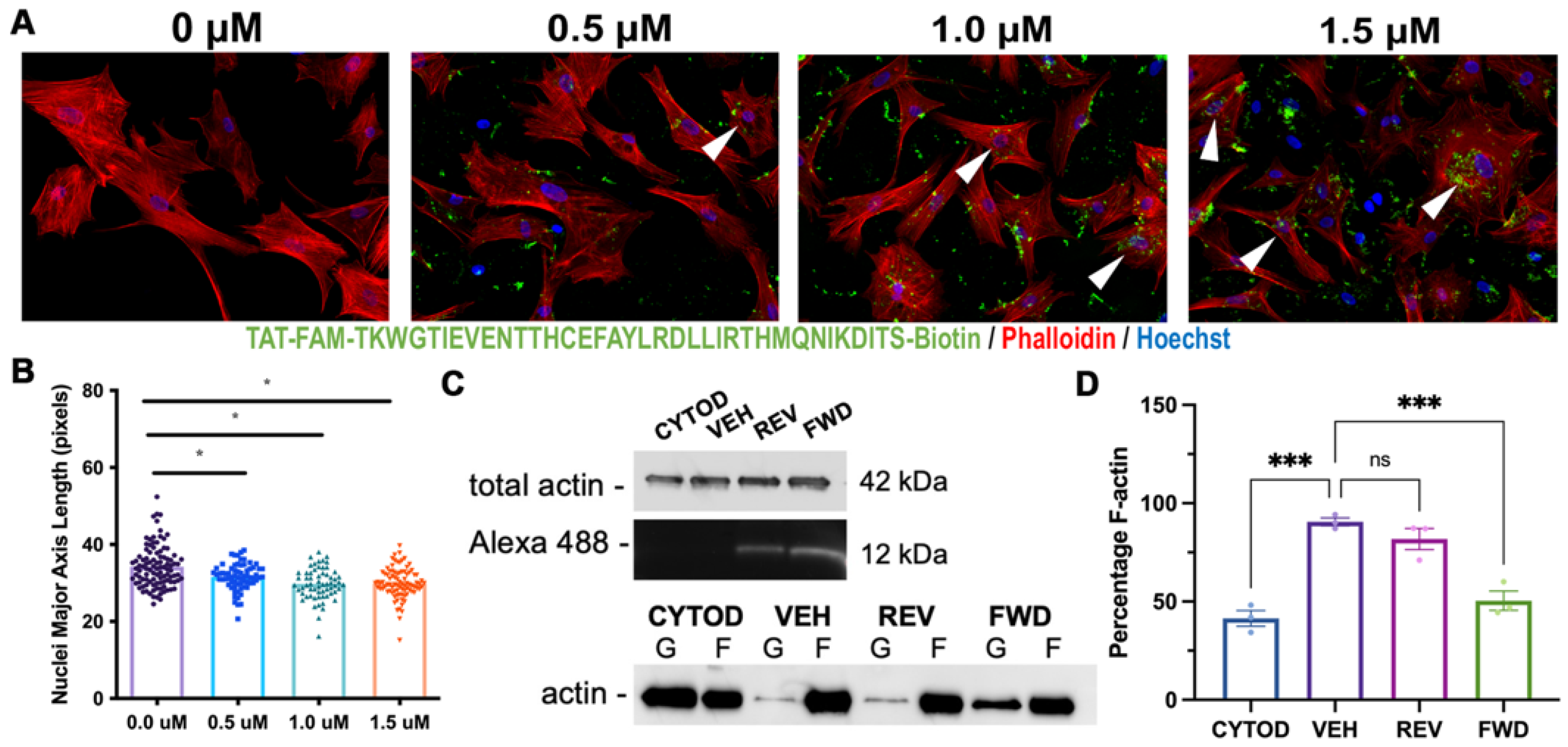
2.3. DCHS1-LIX1L-SEPT9 Is Linked to the Actin Cytoskeleton and Promotes Polymerization
2.4. Consequences of Actin Disorganization in Cells and Tissue
3. Discussion
4. Methods
4.1. Yeast Two-Hybrid Screening Assays
4.2. Co-Immunoprecipatations
4.3. Echocardiography
4.4. Valve Morphometrics
4.5. In Situ Hybridization
4.6. Cell Culture & Treatments
4.7. Immunofluorescence Staining and Imaging
4.8. Quantitative Image Analyses
4.9. Mouse Husbandry and Analyses
4.10. Actin Polymerization Assay
4.11. Transmission Electron Microscopy (TEM)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gammie, J.S.; Chikwe, J.; Badhwar, V.; Thibault, D.P.; Vemulapalli, S.; Thourani, V.H.; Gillinov, M.; Adams, D.H.; Rankin, J.S.; Ghoreishi, M.; et al. Isolated Mitral Valve Surgery: The Society of Thoracic Surgeons Adult Cardiac Surgery Database Analysis. Ann. Thorac. Surg. 2018, 106, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Grigioni, F.; Tribouilloy, C.; Avierinos, J.F.; Barbieri, A.; Ferlito, M.; Trojette, F.; Tafanelli, L.; Branzi, A.; Szymanski, C.; Habib, G.; et al. Outcomes in Mitral Regurgitation Due to Flail Leaflets: A Multicenter European Study. JACC Cardiovasc. Imaging 2008, 1, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Durst, R.; Sauls, K.; Peal, D.S.; DeVlaming, A.; Toomer, K.; Leyne, M.; Salani, M.; Talkowski, M.; Brand, H.; Perrocheau, M.; et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 2015, 525, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Clemenceau, A.; Bérubé, J.-C.; Bélanger, P.; Gaudreault, N.; Lamontagne, M.; Toubal, O.; Clavel, M.-A.; Capoulade, R.; Mathieu, P.; Pibarot, P.; et al. Deleterious variants in DCHS1 are prevalent in sporadic cases of mitral valve prolapse. Mol. Genet. Genom. Med. 2017, 6, 114–120. [Google Scholar] [CrossRef]
- Rogulja, D.; Rauskolb, C.; Irvine, K.D. Morphogen Control of Wing Growth through the Fat Signaling Pathway. Dev. Cell 2008, 15, 309–321. [Google Scholar] [CrossRef]
- Han, M.-R.; Han, K.-M.; Kim, A.; Kang, W.; Kang, Y.; Kang, J.; Won, E.; Tae, W.-S.; Cho, Y.; Ham, B.-J. Whole-exome sequencing identifies variants associated with structural MRI markers in patients with bipolar disorders. J. Affect. Disord. 2019, 249, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Cellini, E.; Vetro, A.; Conti, V.; Marini, C.; Doccini, V.; Clementella, C.; Parrini, E.; Giglio, S.; Della Monica, M.; Fichera, M.; et al. Multiple genomic copy number variants associated with periventricular nodular heterotopia indicate extreme genetic heterogeneity. Eur. J. Hum. Genet. 2019, 27, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Klaus, J.; Kanton, S.; Kyrousi, C.; Ayo-Martin, A.C.; Di Giaimo, R.; Riesenberg, S.; O’Neill, A.C.; Camp, J.G.; Tocco, C.; Santel, M.; et al. Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat. Med. 2019, 25, 561–568. [Google Scholar] [CrossRef]
- Lodge, E.J.; Xekouki, P.; Silva, T.S.; Kochi, C.; Longui, C.A.; Faucz, F.R.; Santambrogio, A.; Mills, J.L.; Pankratz, N.; Lane, J.; et al. Requirement of FAT and DCHS protocadherins during hypothalamic-pituitary development. JCI Insight 2020, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Mulvaney, J.; Zakaria, S.; Yu, T.; Morgan, K.M.; Allen, S.; Basson, M.A.; Francis-West, P.; Irvine, K.D. Characterization of a DCHS1 mutant mouse reveals requirements for DCHS1-Fat4 signaling during mammalian development. Development 2011, 138, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Enriquez, I.; Hodgson, T.; Zakaria, S.; Cadoni, E.; Shah, M.; Allen, S.; Al-Khishali, A.; Mao, Y.; Yiu, A.; Petzold, J.; et al. DCHS1-Fat4 regulation of osteogenic differentiation in mouse. Development 2019, 146, 146. [Google Scholar] [CrossRef]
- Mao, Y.; Kuta, A.; Crespo-Enriquez, I.; Whiting, D.; Martin, T.; Mulvaney, J.; Irvine, K.D.; Francis-West, P. DCHS1–Fat4 regulation of polarized cell behaviours during skeletal morphogenesis. Nat. Commun. 2016, 7, 11469. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Irvine, K.D. Action of fat, four-jointed, dachsous and dachs in distal-to-proximal wing signaling. Development 2004, 131, 4489–4500. [Google Scholar] [CrossRef]
- Mao, Y.; Rauskolb, C.; Cho, E.; Hu, W.-L.; Hayter, H.; Minihan, G.; Katz, F.N.; Irvine, K.D. Dachs: An unconventional myosin that functions downstream of Fat to regulate growth, affinity and gene expression in Drosophila. Development 2006, 133, 2539–2551. [Google Scholar] [CrossRef] [PubMed]
- Bosveld, F.; Bonnet, I.; Guirao, B.; Tlili, S.; Wang, Z.; Petitalot, A.; Marchand, R.; Bardet, P.-L.; Marcq, P.; Graner, F.; et al. Mechanical Control of Morphogenesis by Fat/Dachsous/Four-Jointed Planar Cell Polarity Pathway. Science 2012, 336, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, X.; Zhang, M.; Zhang, Y.; Ye, S.; Leng, Y.; Yang, T.; Kong, L.; Zhang, H. Limb expression 1-like (LIX1L) protein promotes cholestatic liver injury by regulating bile acid metabolism. J. Hepatol. 2021, 75, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Kahyo, T.; Tao, H.; Shibata, K.; Kurabe, N.; Yamada, H.; Shinmura, K.; Ohnishi, K.; Sugimura, H. Novel roles for LIX1L in promoting cancer cell proliferation through ROS1-mediated LIX1L phosphorylation. Sci. Rep. 2015, 5, 13474. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iliadis, C.; Baldus, S.; Kalbacher, D.; Boekstegers, P.; Schillinger, W.; Ouarrak, T.; Zahn, R.; Butter, C.; Zuern, C.S.; Von Bardeleben, R.S.; et al. Impact of left atrial diameter on outcome in patients undergoing edge-to-edge mitral valve repair: Results from the German TRAnscatheter Mitral valve Interventions (TRAMI) registry. Eur. J. Heart Fail. 2020, 22, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Cameli, M.; Incampo, E.; Mondillo, S. Left atrial deformation: Useful index for early detection of cardiac damage in chronic mitral regurgitation. IJC Heart Vasc. 2017, 17, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Schoen, F.J. Calcific and Degenerative Heart Valve Disease. In Cellular and Molecular Pathobiology of Cardiovascular Disease; Academic Press: Amsterdam, The Netherlands, 2014; pp. 161–180. [Google Scholar] [CrossRef]
- Neri, T.; Hiriart, E.; van Vliet, P.; Faure, E.; Norris, R.A.; Farhat, B.; Jagla, B.; Lefrancois, J.; Sugi, Y.; Moore-Morris, T.; et al. Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat. Commun. 2019, 10, 1929. [Google Scholar] [CrossRef] [PubMed]
- Mostowy, S.; Cossart, P. Septins: The fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 183–194. [Google Scholar] [CrossRef]
- Dolat, L.; Hu, Q.; Spiliotis, E.T. Septin functions in organ system physiology and pathology. Biol. Chem. 2013, 395, 123–141. [Google Scholar] [CrossRef]
- Moore, K.; Moore, R.; Wang, C.; Norris, R.A. Tugging at the Heart Strings: The Septin Cytoskeleton in Heart Development and Disease. J. Cardiovasc. Dev. Dis. 2020, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Versaevel, M.; Grevesse, T.; Gabriele, S. Spatial coordination between cell and nuclear shape within micropatterned endothelial cells. Nat. Commun. 2012, 3, 671. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Schulze, N.; Graessl, M.; Soares, A.B.; Geyer, M.; Dehmelt, L.; Nalbant, P. FHOD1 regulates stress fiber organization by controlling transversal arc and dorsal fiber dynamics. J. Cell Sci. 2014, 127, 1379–1393. [Google Scholar] [CrossRef]
- Dolat, L.; Hunyara, J.L.; Bowen, J.R.; Karasmanis, E.; Elgawly, M.; Galkin, V.E.; Spiliotis, E.T. Septins promote stress fiber–mediated maturation of focal adhesions and renal epithelial motility. J. Cell Biol. 2014, 207, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B., Jr.; Lincoln, J.; Deutsch, G.H.; Osinska, H.; Manning, P.B.; Benson, D.W.; Yutzey, K.E. Extracellular Matrix Remodeling and Organization in Developing and Diseased Aortic Valves. Circ. Res. 2006, 98, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Toomer, K.A.; Yu, M.; Fulmer, D.; Guo, L.; Moore, K.S.; Moore, R.; Drayton, K.D.; Glover, J.; Peterson, N.; Ramos-Ortiz, S.; et al. Primary cilia defects causing mitral valve prolapse. Sci. Transl. Med. 2019, 11, 11. [Google Scholar] [CrossRef]
- Fulmer, D.; Toomer, K.A.; Glover, J.; Guo, L.; Moore, K.; Moore, R.; Stairley, R.; Gensemer, C.; Abrol, S.; Rumph, M.K.; et al. Desert hedgehog-primary cilia cross talk shapes mitral valve tissue by organizing smooth muscle actin. Dev. Biol. 2020, 463, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Fulmer, D.; Guo, L.; Koren, N.; Glover, J.; Moore, R.; Gensemer, C.; Beck, T.; Morningstar, J.; Stairley, R.; et al. PDGFRα: Expression and Function during Mitral Valve Morphogenesis. J. Cardiovasc. Dev. Dis. 2021, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Beck, T.; Fulmer, D.; Ramos-Ortiz, S.; Glover, J.; Wang, C.; Moore, K.; Gensemer, C.; Morningstar, J.; Moore, R.; et al. DZIP1 regulates mammalian cardiac valve development through a Cby1-beta-catenin mechanism. Dev. Dyn. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sauls, K.; de Vlaming, A.; Harris, B.S.; Williams, K.; Wessels, A.; Levine, R.A.; Slaugenhaupt, S.A.; Goodwin, R.L.; Pavone, L.M.; Merot, J.; et al. Developmental basis for filamin-A-associated myxomatous mitral valve disease. Cardiovasc. Res. 2012, 96, 109–119. [Google Scholar] [CrossRef]
- Dina, C.; PROMESA Investigators; Bouatia-Naji, N.; Tucker, N.; Delling, F.N.; Toomer, K.; Durst, R.; Perrocheau, M.; Fernandez-Friera, L.; Solis, J.; et al. Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat. Genet. 2015, 47, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Spiliotis, E.T.; Nakos, K. Cellular functions of actin- and microtubule-associated septins. Curr. Biol. 2021, 31, R651–R666. [Google Scholar] [CrossRef]
- Butcher, J.T.; McQuinn, T.C.; Sedmera, D.; Turner, D.; Markwald, R.R. Transitions in Early Embryonic Atrioventricular Valvular Function Correspond with Changes in Cushion Biomechanics That Are Predictable by Tissue Composition. Circ. Res. 2007, 100, 1503–1511. [Google Scholar] [CrossRef]
- Butcher, J.T.; Nerem, R.M. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos. Trans. R. Soc. B Biol. Sci. 2007, 362, 1445–1457. [Google Scholar] [CrossRef]
- Yalcin, H.C.; Shekhar, A.; McQuinn, T.C.; Butcher, J.T. Hemodynamic patterning of the avian atrioventricular valve. Dev. Dyn. 2011, 240, 23–35. [Google Scholar] [CrossRef]
- Gould, R.A.; Yalcin, H.C.; MacKay, J.L.; Sauls, K.; Norris, R.; Kumar, S.; Butcher, J.T. Cyclic Mechanical Loading Is Essential for Rac1-Mediated Elongation and Remodeling of the Embryonic Mitral Valve. Curr. Biol. 2016, 26, 27–37. [Google Scholar] [CrossRef]
- De Vlaming, A.; Sauls, K.; Hajdu, Z.; Visconti, R.P.; Mehesz, A.N.; Levine, R.A.; Slaugenhaupt, S.A.; Hagège, A.; Chester, A.H.; Markwald, R.R.; et al. Atrioventricular valve development: New perspectives on an old theme. Differentiation 2012, 84, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Gloerich, M.; Bianchini, J.M.; Siemers, K.A.; Cohen, D.J.; Nelson, W.J. Cell division orientation is coupled to cell–cell adhesion by the E-cadherin/LGN complex. Nat. Commun. 2017, 8, 13996. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Pagnozzi, L.A.; Butcher, J.T. Mechanotransduction Mechanisms in Mitral Valve Physiology and Disease Pathogenesis. Front. Cardiovasc. Med. 2017, 4, 83. [Google Scholar] [CrossRef]
- Le Duc, Q.; Shi, Q.; Blonk, I.; Sonnenberg, A.; Wang, N.; Leckband, D.; de Rooij, J. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II–dependent manner. J. Cell Biol. 2010, 189, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Cao, Y.; Liu, L.; Zhao, J.; Zhang, T.; Xiao, L.; Jia, M.; Tian, Q.; Yu, H.; Chen, S.; et al. SEPT9_i1 regulates human breast cancer cell motility through cytoskeletal and RhoA/FAK signaling pathway regulation. Cell Death Dis. 2019, 10, 720. [Google Scholar] [CrossRef] [PubMed]
- Marcus, J.; Bejerano-Sagie, M.; Patterson, N.; Bagchi, S.; Verkhusha, V.; Connolly, D.; Goldberg, G.L.; Golden, A.; Sharma, V.P.; Condeelis, J.; et al. Septin 9 isoforms promote tumorigenesis in mammary epithelial cells by increasing migration and ECM degradation through metalloproteinase secretion at focal adhesions. Oncogene 2019, 38, 5839–5859. [Google Scholar] [CrossRef]
- Fierro-González, J.C.; White, M.D.; Silva, J.C.; Plachta, N. Cadherin-dependent filopodia control preimplantation embryo compaction. Nat. Cell Biol. 2013, 15, 1424–1433. [Google Scholar] [CrossRef]
- Pujol, F.; Hodgson, T.; Martinez-Corral, I.; Prats, A.-C.; Devenport, D.; Takeichi, M.; Genot, E.; Mäkinen, T.; Francis-West, P.; Garmy-Susini, B.; et al. Dachsous1–Fat4 Signaling Controls Endothelial Cell Polarization During Lymphatic Valve Morphogenesis—Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1732–1735. [Google Scholar] [CrossRef]
- Casas-Tintó, S.; Portela, M. Cytonemes, Their Formation, Regulation, and Roles in Signaling and Communication in Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 5641. [Google Scholar] [CrossRef]
- Bodeen, W.J.; Marada, S.; Truong, A.; Ogden, S.K. A fixation method to preserve cultured cell cytonemes facilitates mechanistic interrogation of morphogen transport. Development 2017, 144, 3612–3624. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.T.; Dillard, M.E.; Stewart, D.P.; Zhang, Y.; Wagner, B.; Levine, R.M.; Pruett-Miller, S.M.; Sykes, A.; Temirov, J.; Cheney, R.E.; et al. Cytoneme delivery of Sonic Hedgehog from ligand-producing cells requires Myosin 10 and a Dispatched-BOC/CDON co-receptor complex. eLife 2021, 10, e61432. [Google Scholar] [CrossRef]
- Brunt, L.; Greicius, G.; Rogers, S.; Evans, B.D.; Virshup, D.M.; Wedgwood, K.C.A.; Scholpp, S. Vangl2 promotes the formation of long cytonemes to enable distant Wnt/β-catenin signaling. Nat. Commun. 2021, 12, 2058. [Google Scholar] [CrossRef] [PubMed]
- Rao-Bhatia, A.; Zhu, M.; Yin, W.-C.; Coquenlorge, S.; Zhang, X.; Woo, J.; Sun, Y.; Dean, C.H.; Liu, A.; Hui, C.-C.; et al. Hedgehog-Activated Fat4 and PCP Pathways Mediate Mesenchymal Cell Clustering and Villus Formation in Gut Development. Dev. Cell 2020, 52, 647–658.e6. [Google Scholar] [CrossRef] [PubMed]
- Dau, C.; Fliegauf, M.; Omran, H.; Schlensog, M.; Dahl, E.; van Roeyen, C.R.; Kriz, W.; Moeller, M.J.; Braun, G.S. The atypical cadherin Dachsous1 localizes to the base of the ciliary apparatus in airway epithelia. Biochem. Biophys. Res. Commun. 2016, 473, 1177–1184. [Google Scholar] [CrossRef]
- Li-Villarreal, N.; Forbes, M.M.; Loza, A.J.; Chen, J.; Ma, T.; Helde, K.; Moens, C.B.; Shin, J.; Sawada, A.; Hindes, A.E.; et al. Dachsous1b cadherin regulates actin and microtubule cytoskeleton during early zebrafish embryogenesis. Development 2015, 142, 2704–2718. [Google Scholar] [CrossRef]
- Chen, J.; Castelvecchi, G.D.; Li-Villarreal, N.; Raught, B.; Krezel, A.M.; McNeill, H.; Solnica-Krezel, L. Atypical Cadherin Dachsous1b Interacts with Ttc28 and Aurora B to Control Microtubule Dynamics in Embryonic Cleavages. Dev. Cell 2018, 45, 376–391.e5. [Google Scholar] [CrossRef]
- Ghossoub, R.; Hu, Q.; Failler, M.; Rouyez, M.-C.; Spitzbarth, B.; Mostowy, S.; Wolfrum, U.; Saunier, S.; Cossart, P.; Nelson, W.J.; et al. Septins 2, 7, and 9 and MAP4 co-localize along the axoneme in the primary cilium and control ciliary length. J. Cell Sci. 2013, 126, 2583–2594. [Google Scholar] [CrossRef]
- Toomer, K.A.; Fulmer, D.; Guo, L.; Drohan, A.; Peterson, N.; Swanson, P.; Brooks, B.; Mukherjee, R.; Body, S.; Lipschutz, J.H.; et al. A role for primary cilia in aortic valve development and disease. Dev. Dyn. 2017, 246, 625–634. [Google Scholar] [CrossRef]
- Fulmer, D.; Toomer, K.; Guo, L.; Moore, K.; Glover, J.; Moore, R.; Stairley, R.; Lobo, G.; Zuo, X.; Dang, Y.; et al. Defects in the Exocyst-Cilia Machinery Cause Bicuspid Aortic Valve Disease and Aortic Stenosis. Circulation 2019, 140, 1331–1341. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, K.S.; Moore, R.; Fulmer, D.B.; Guo, L.; Gensemer, C.; Stairley, R.; Glover, J.; Beck, T.C.; Morningstar, J.E.; Biggs, R.; et al. DCHS1, Lix1L, and the Septin Cytoskeleton: Molecular and Developmental Etiology of Mitral Valve Prolapse. J. Cardiovasc. Dev. Dis. 2022, 9, 62. https://doi.org/10.3390/jcdd9020062
Moore KS, Moore R, Fulmer DB, Guo L, Gensemer C, Stairley R, Glover J, Beck TC, Morningstar JE, Biggs R, et al. DCHS1, Lix1L, and the Septin Cytoskeleton: Molecular and Developmental Etiology of Mitral Valve Prolapse. Journal of Cardiovascular Development and Disease. 2022; 9(2):62. https://doi.org/10.3390/jcdd9020062
Chicago/Turabian StyleMoore, Kelsey S., Reece Moore, Diana B. Fulmer, Lilong Guo, Cortney Gensemer, Rebecca Stairley, Janiece Glover, Tyler C. Beck, Jordan E. Morningstar, Rachel Biggs, and et al. 2022. "DCHS1, Lix1L, and the Septin Cytoskeleton: Molecular and Developmental Etiology of Mitral Valve Prolapse" Journal of Cardiovascular Development and Disease 9, no. 2: 62. https://doi.org/10.3390/jcdd9020062
APA StyleMoore, K. S., Moore, R., Fulmer, D. B., Guo, L., Gensemer, C., Stairley, R., Glover, J., Beck, T. C., Morningstar, J. E., Biggs, R., Muhkerjee, R., Awgulewitsch, A., & Norris, R. A. (2022). DCHS1, Lix1L, and the Septin Cytoskeleton: Molecular and Developmental Etiology of Mitral Valve Prolapse. Journal of Cardiovascular Development and Disease, 9(2), 62. https://doi.org/10.3390/jcdd9020062