
In Vivo Methods to Monitor Cardiomyocyte Proliferation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
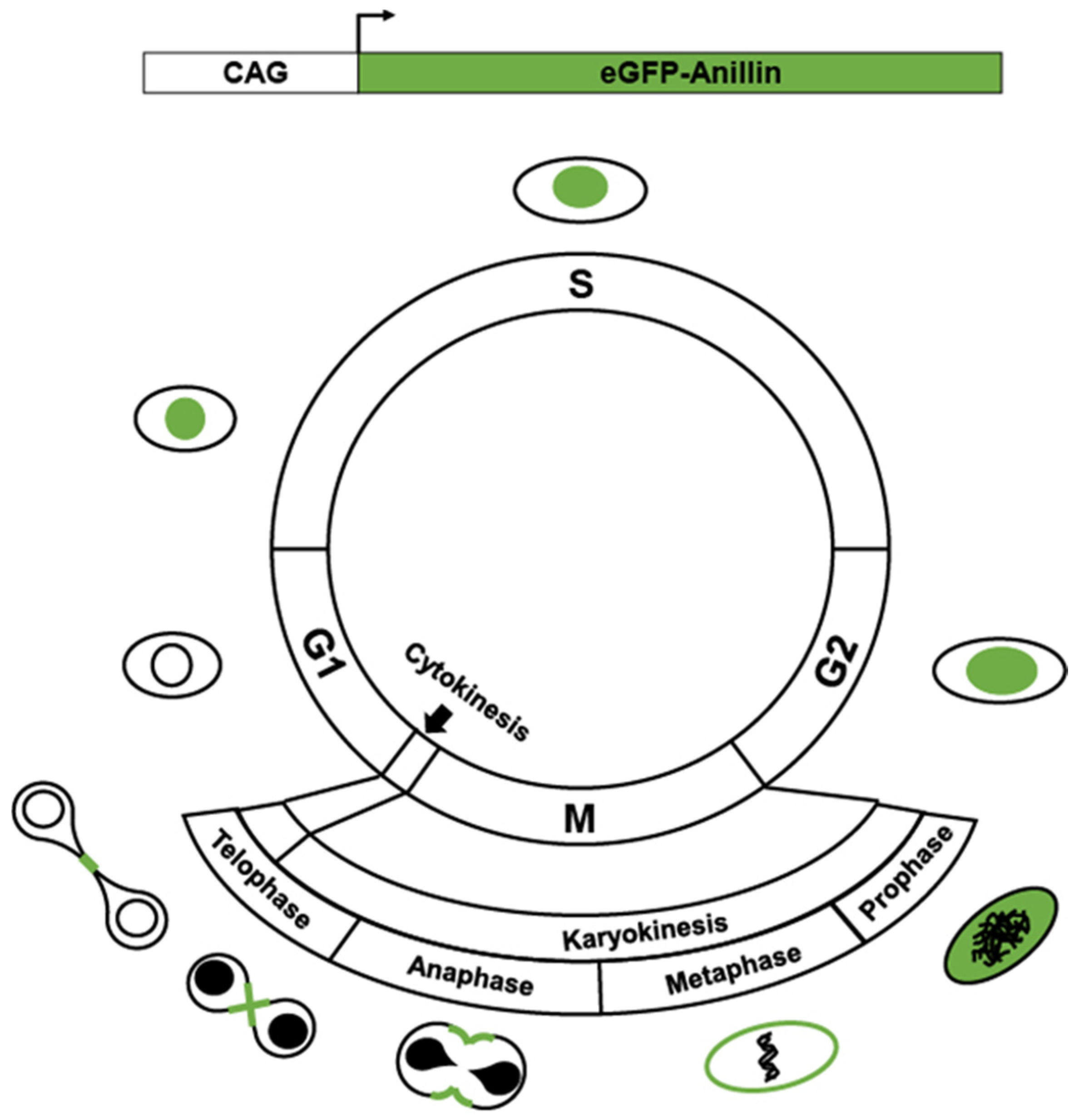{kind=link}
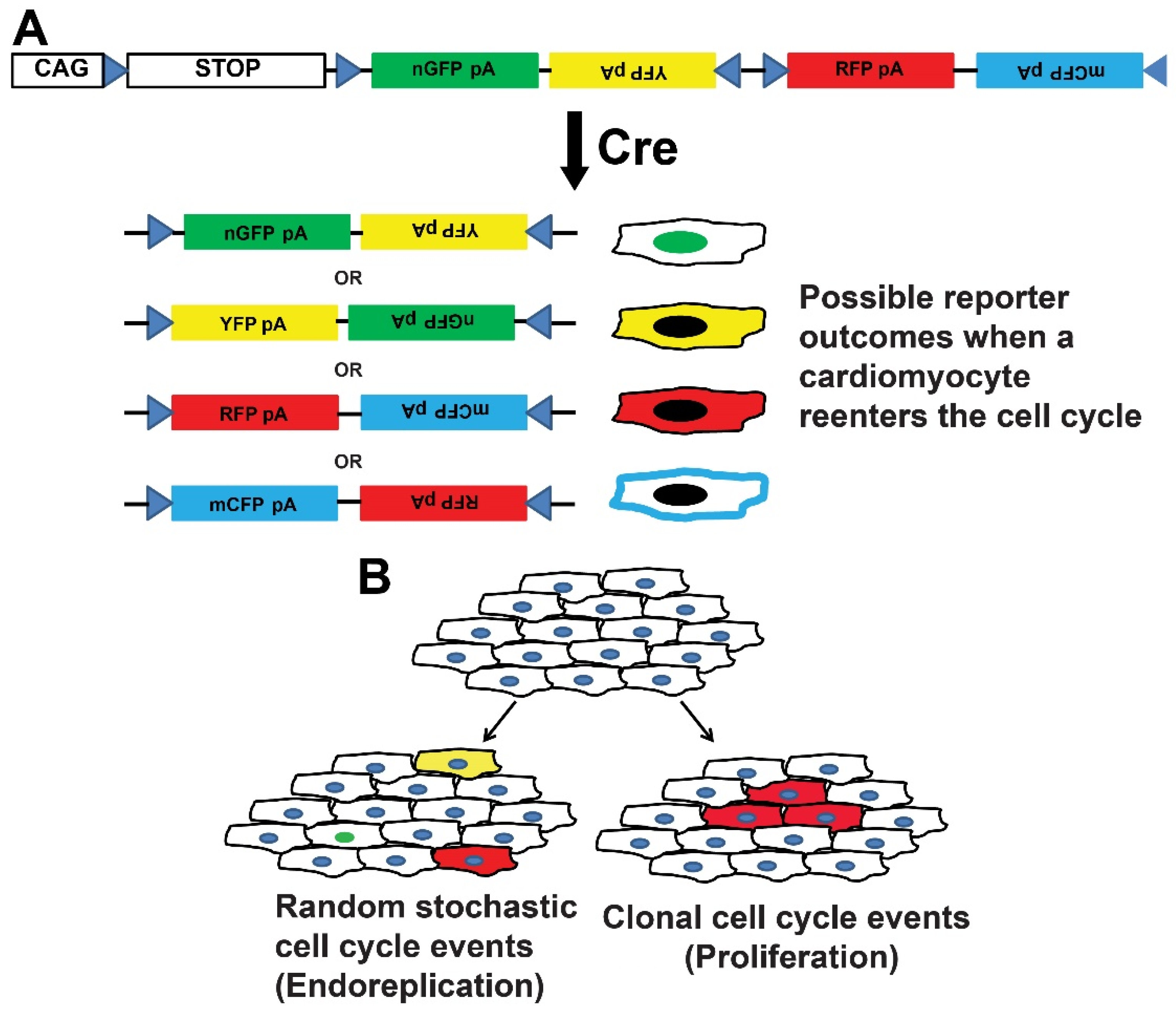{kind=link}
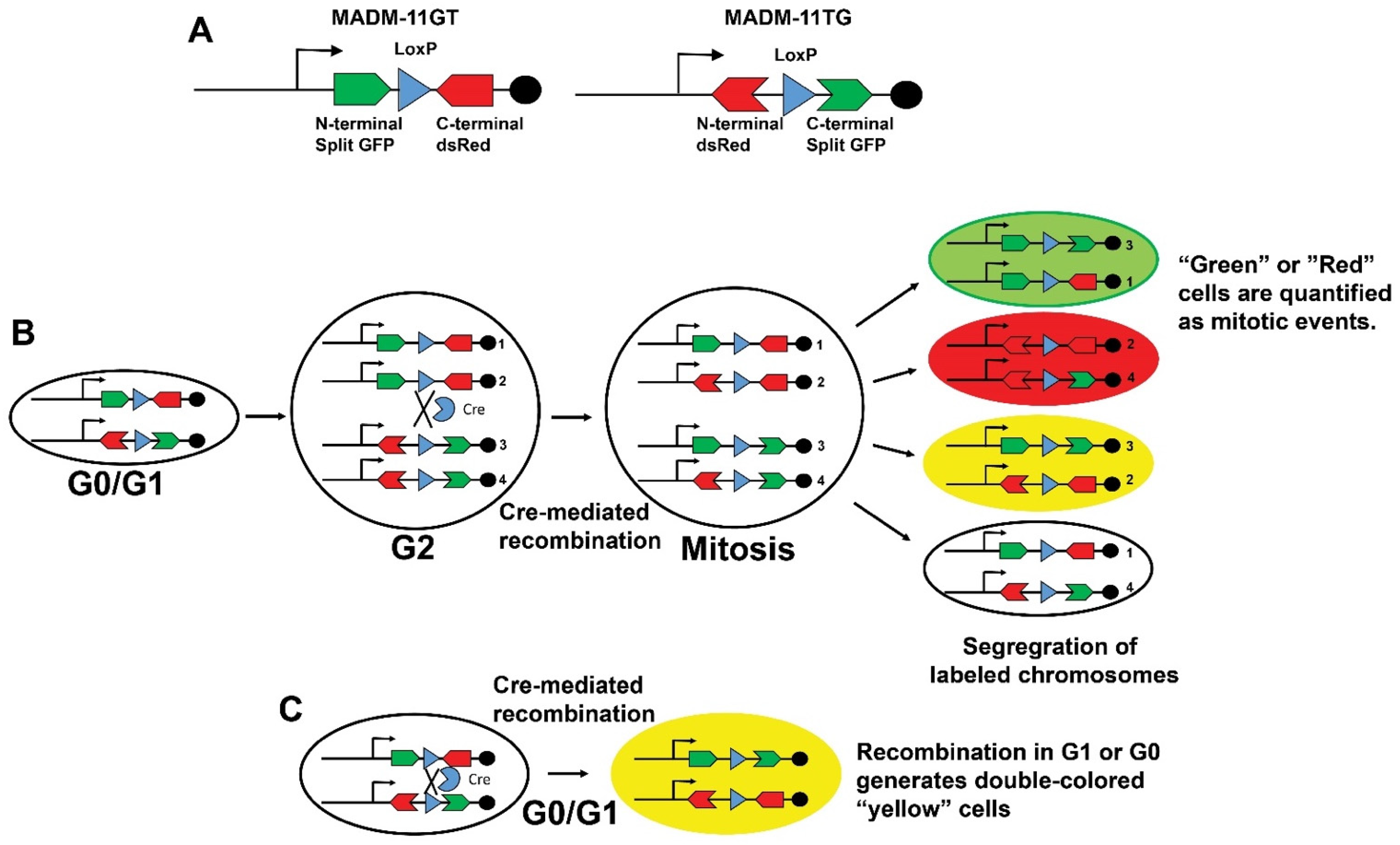{kind=link}
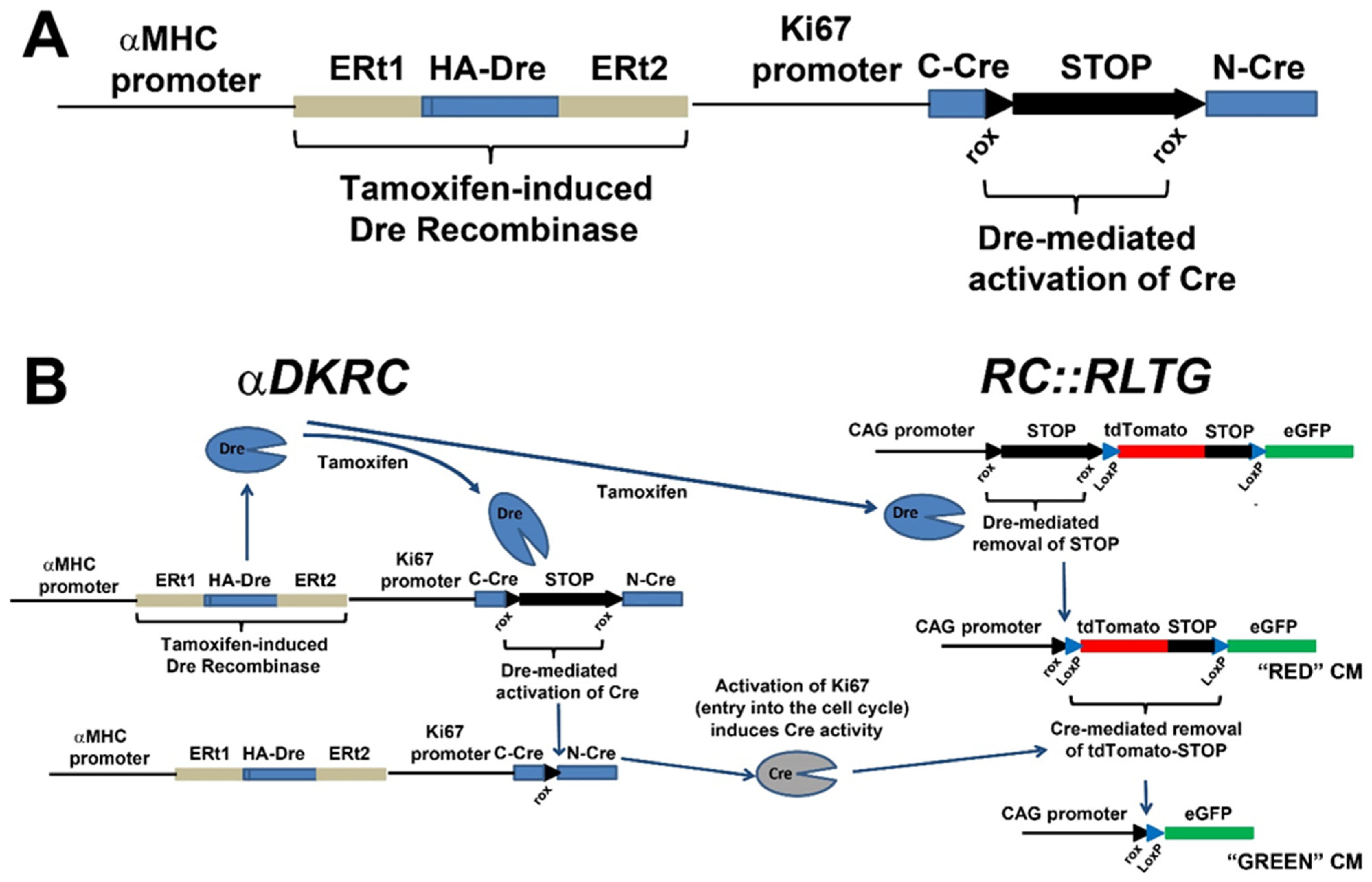{kind=link}
Abstract
:1. Introduction
1.1. Normal Mammalian Heart Cycling, Exit from the Cell Cycle, and Estimates of Cardiomyocyte Turnover in Human Hearts
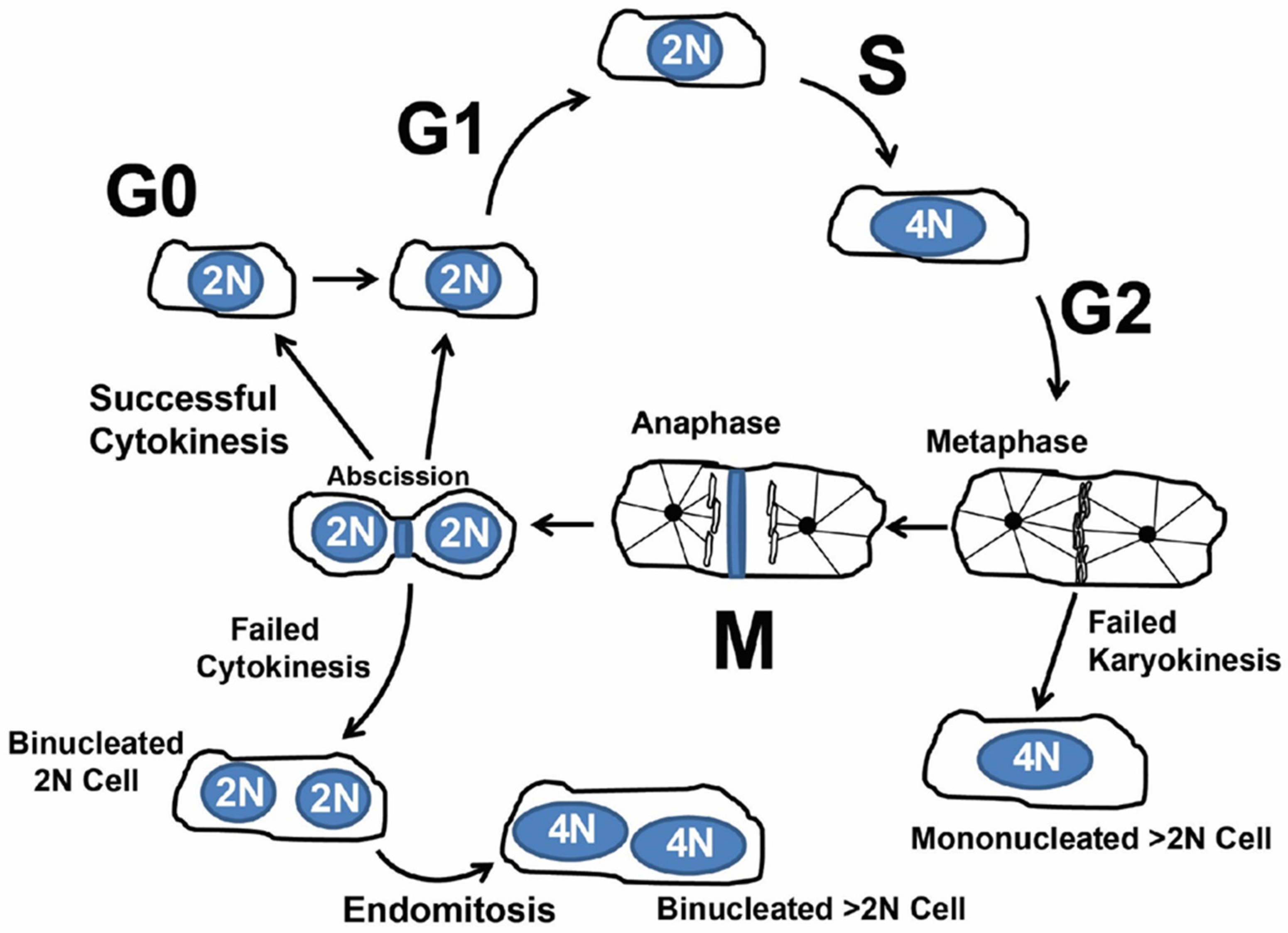
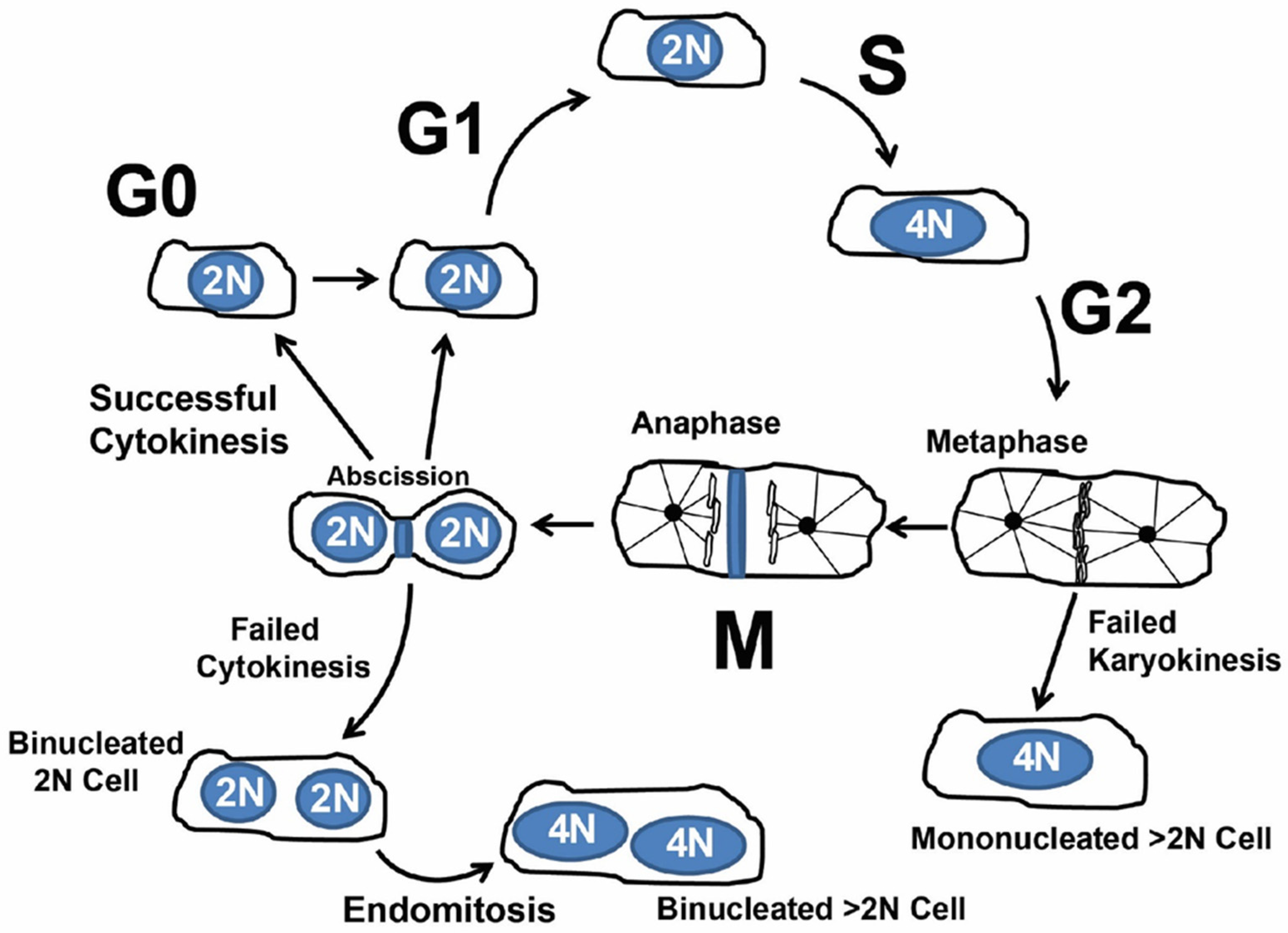
1.2. Cardiomyocyte Endoreplication and Binucleation vs. Proliferation
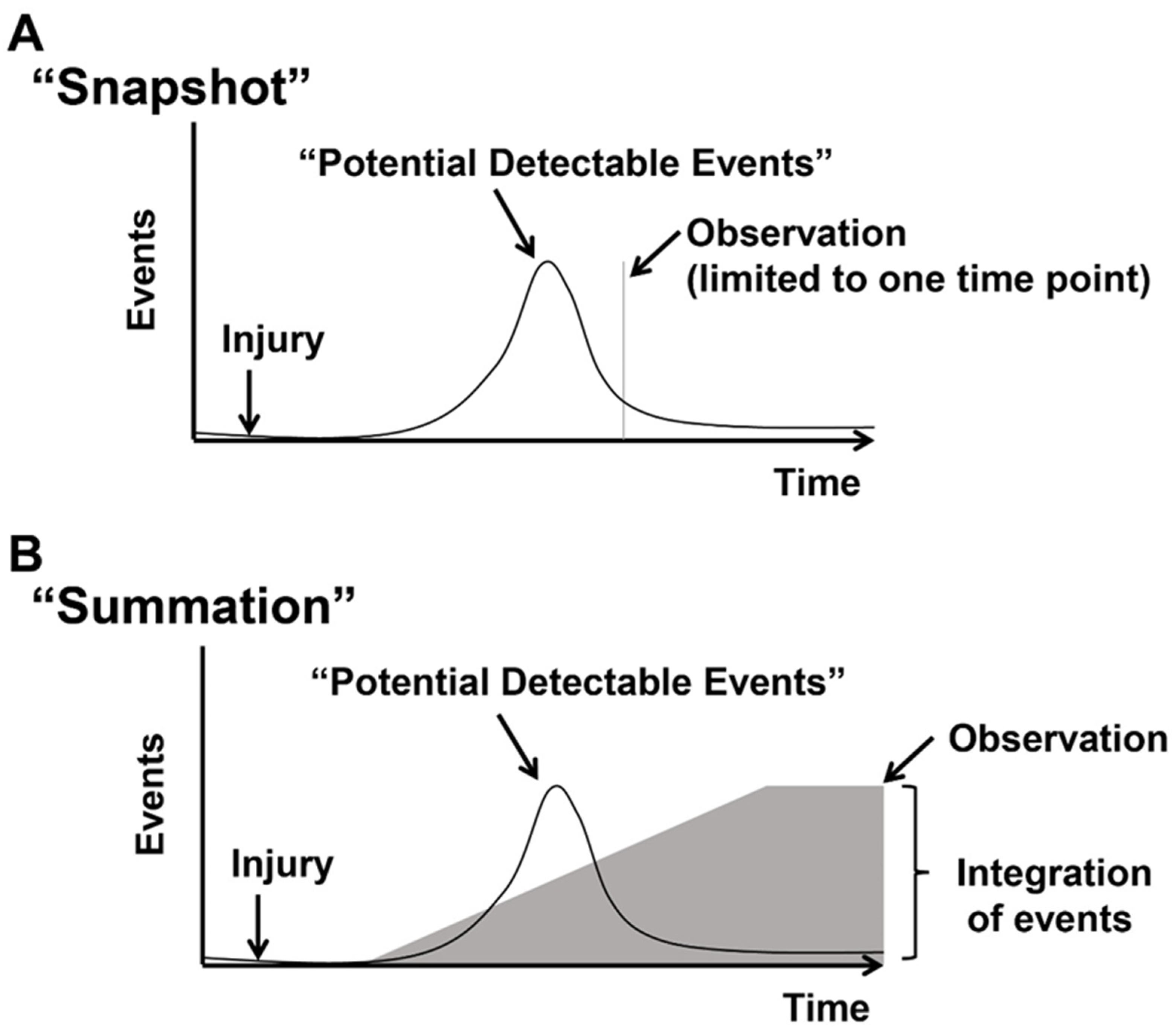
1.3. Concept of Actively Cycling and Previously Cycled Cardiomyocytes and Overview of “Snapshot” vs. “Integration of Events” Using an Indelible Mark
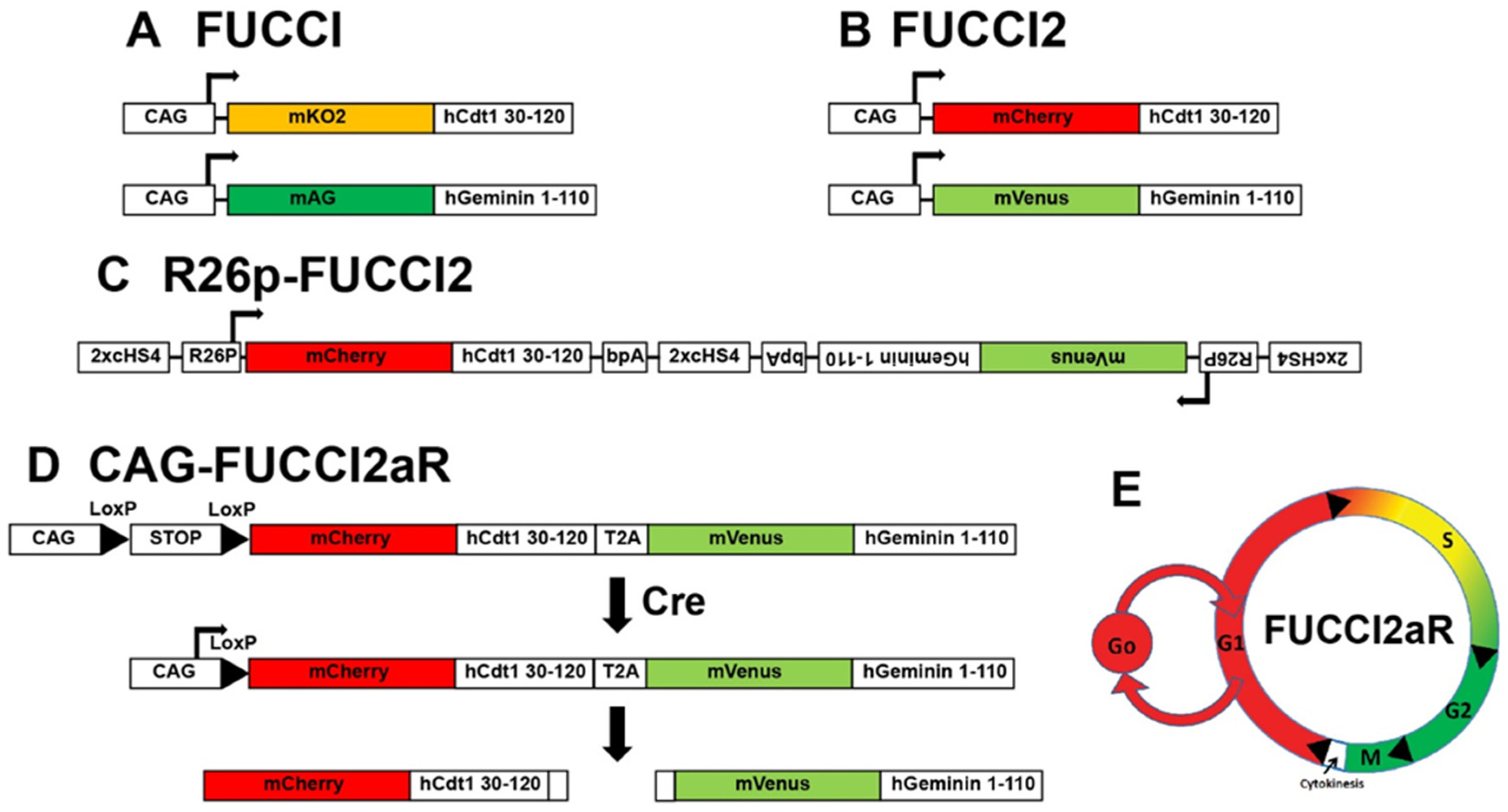
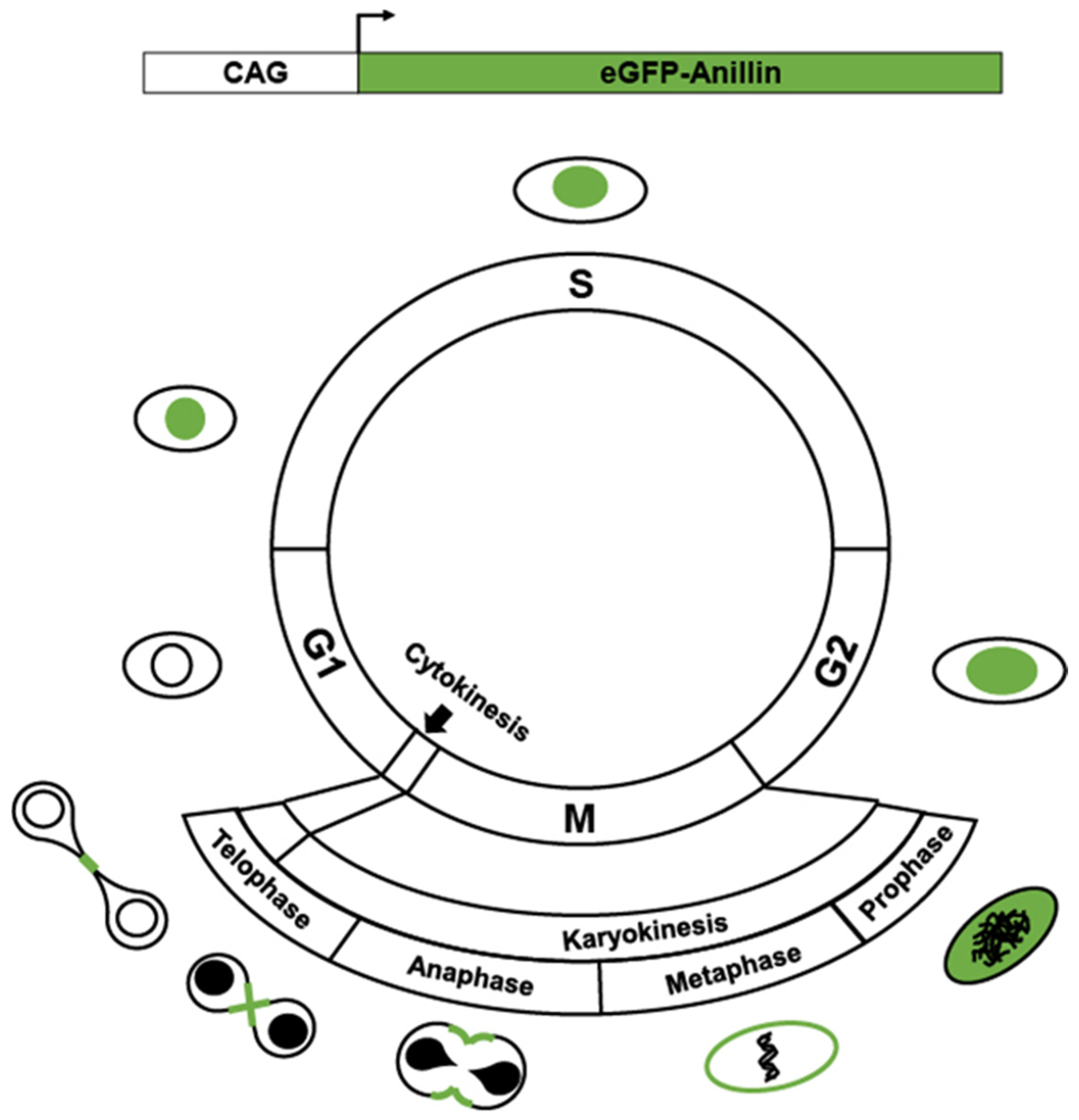
2. Transgenics Reporter Mice of Actively Cycling Cells
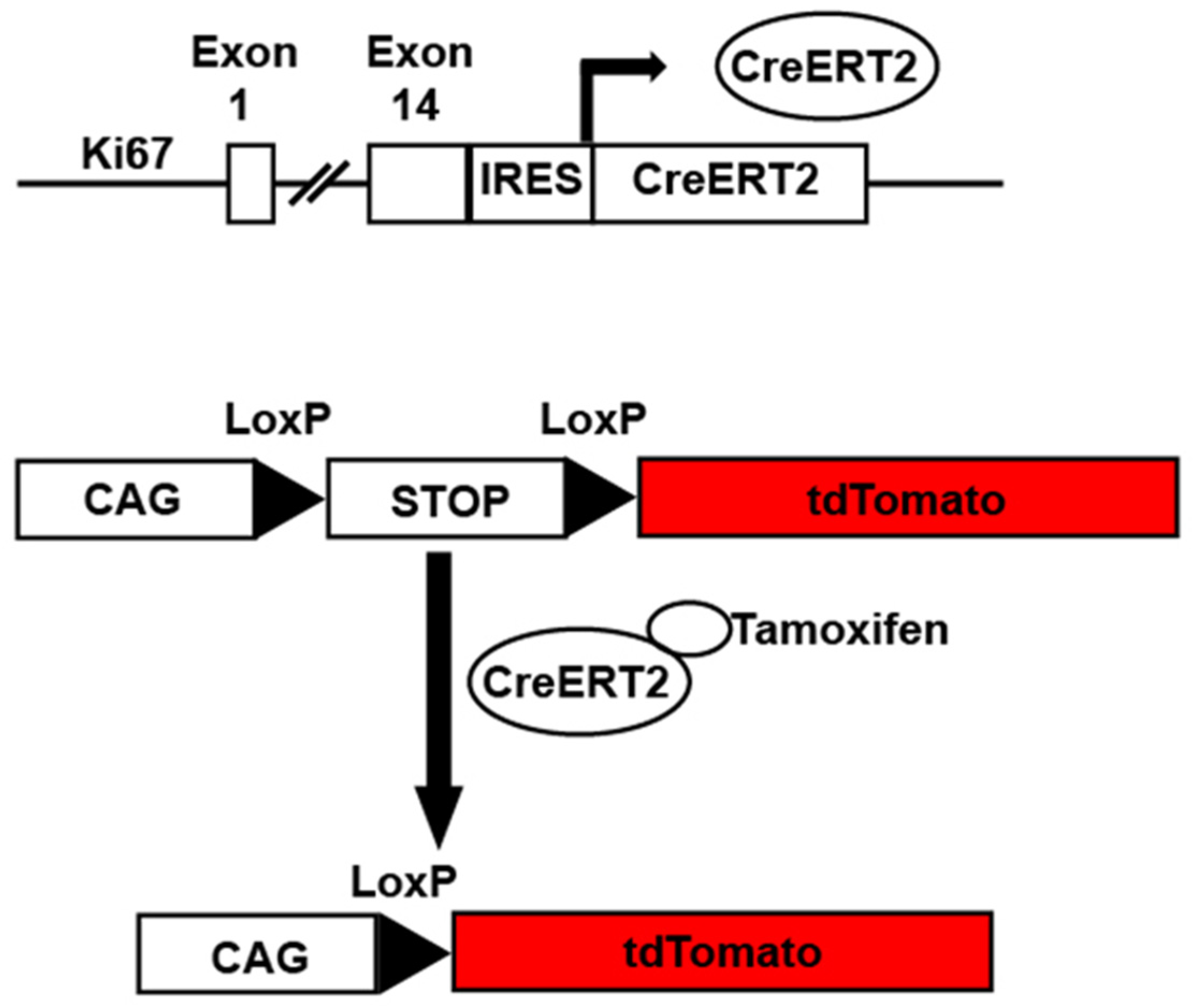
3. Transgenic Reporter Mice of Previously Cycled Cells
4. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef] [PubMed]
- Eschenhagen, T.; Bolli, R.; Braun, T.; Field, L.J.; Fleischmann, B.K.; Frisen, J.; Giacca, M.; Hare, J.M.; Houser, S.; Lee, R.T.; et al. Cardiomyocyte Regeneration: A Consensus Statement. Circulation 2017, 136, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Alkass, K.; Panula, J.; Westman, M.; Wu, T.D.; Guerquin-Kern, J.L.; Bergmann, O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell 2015, 163, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Bradley, L.A.; Young, A.; Li, H.; Billcheck, H.O.; Wolf, M.J. Loss of Endogenously Cycling Adult Cardiomyocytes Worsens Myocardial Function. Circ. Res. 2021, 128, 155–168. [Google Scholar] [CrossRef]
- D’Uva, G.; Aharonov, A.; Lauriola, M.; Kain, D.; Yahalom-Ronen, Y.; Carvalho, S.; Weisinger, K.; Bassat, E.; Rajchman, D.; Yifa, O.; et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015, 17, 627–638. [Google Scholar] [CrossRef]
- Hesse, M.; Doengi, M.; Becker, A.; Kimura, K.; Voeltz, N.; Stein, V.; Fleischmann, B.K. Midbody Positioning and Distance between Daughter Nuclei Enable Unequivocal Identification of Cardiomyocyte Cell Division in Mice. Circ. Res. 2018, 123, 1039–1052. [Google Scholar] [CrossRef]
- Lin, Z.; von Gise, A.; Zhou, P.; Gu, F.; Ma, Q.; Jiang, J.; Yau, A.L.; Buck, J.N.; Gouin, K.A.; van Gorp, P.R.; et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res. 2014, 115, 354–363. [Google Scholar] [CrossRef]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Sereti, K.I.; Nguyen, N.B.; Kamran, P.; Zhao, P.; Ranjbarvaziri, S.; Park, S.; Sabri, S.; Engel, J.L.; Sung, K.; Kulkarni, R.P.; et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat. Commun. 2018, 9, 754. [Google Scholar] [CrossRef]
- Soonpaa, M.H.; Kim, K.K.; Pajak, L.; Franklin, M.; Field, L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 1996, 271, H2183–H2189. [Google Scholar] [CrossRef]
- Soonpaa, M.H.; Koh, G.Y.; Pajak, L.; Jing, S.; Wang, H.; Franklin, M.T.; Kim, K.K.; Field, L.J. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J. Clin. Investig. 1997, 99, 2644–2654. [Google Scholar] [CrossRef] [Green Version]
- Derks, W.; Bergmann, O. Polyploidy in Cardiomyocytes: Roadblock to Heart Regeneration? Circ. Res. 2020, 126, 552–565. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Lazar, E.; Sadek, H.A.; Bergmann, O. Cardiomyocyte renewal in the human heart: Insights from the fall-out. Eur. Heart J. 2017, 38, 2333–2342. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Zdunek, S.; Alkass, K.; Druid, H.; Bernard, S.; Frisen, J. Identification of cardiomyocyte nuclei and assessment of ploidy for the analysis of cell turnover. Exp. Cell Res. 2011, 317, 188–194. [Google Scholar] [CrossRef]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar]
- Gerdes, J.; Schwab, U.; Lemke, H.; Stein, H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int. J. Cancer 1983, 31, 13–20. [Google Scholar] [CrossRef]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Broad, A.J.; DeLuca, J.G. The right place at the right time: Aurora B kinase localization to centromeres and kinetochores. Essays Biochem. 2020, 64, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bian, M.; Jiang, Q.; Zhang, C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol. Cancer Res. 2007, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hans, F.; Dimitrov, S. Histone H3 phosphorylation and cell division. Oncogene 2001, 20, 3021–3027. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Yu, L.; Bowen, J.; Gorovsky, M.A.; Allis, C.D. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 1999, 97, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Carim, S.C.; Kechad, A.; Hickson, G.R.X. Animal Cell Cytokinesis: The Rho-Dependent Actomyosin-Anilloseptin Contractile Ring as a Membrane Microdomain Gathering, Compressing, and Sorting Machine. Front. Cell Dev. Biol. 2020, 8, 575226. [Google Scholar] [CrossRef]
- Piekny, A.J.; Maddox, A.S. The myriad roles of Anillin during cytokinesis. Semin. Cell Dev. Biol. 2010, 21, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Maddox, A.S. Anillin. Curr. Biol. 2010, 20, R135–R136. [Google Scholar] [CrossRef] [Green Version]
- Heallen, T.; Morikawa, Y.; Leach, J.; Tao, G.; Willerson, J.T.; Johnson, R.L.; Martin, J.F. Hippo signaling impedes adult heart regeneration. Development 2013, 140, 4683–4690. [Google Scholar] [CrossRef] [Green Version]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef]
- Hirose, K.; Payumo, A.Y.; Cutie, S.; Hoang, A.; Zhang, H.; Guyot, R.; Lunn, D.; Bigley, R.B.; Yu, H.; Wang, J.; et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 2019, 364, 184–188. [Google Scholar] [CrossRef]
- Soonpaa, M.H.; Field, L.J. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am. J. Physiol. 1997, 272, H220–H226. [Google Scholar] [CrossRef]
- Katz, E.B.; Steinhelper, M.E.; Delcarpio, J.B.; Daud, A.I.; Claycomb, W.C.; Field, L.J. Cardiomyocyte proliferation in mice expressing alpha-cardiac myosin heavy chain-SV40 T-antigen transgenes. Am. J. Physiol. 1992, 262, H1867–H1876. [Google Scholar] [CrossRef]
- Pasumarthi, K.B.; Nakajima, H.; Nakajima, H.O.; Soonpaa, M.H.; Field, L.J. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ. Res. 2005, 96, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Soonpaa, M.H.; Zebrowski, D.C.; Platt, C.; Rosenzweig, A.; Engel, F.B.; Field, L.J. Cardiomyocyte Cell-Cycle Activity during Preadolescence. Cell 2015, 163, 781–782. [Google Scholar] [CrossRef] [Green Version]
- Brodskiǐ, V.I.A.; Uryvaeva, I.V.; Brodskiǐ, V.I.A. Genome Multiplication in Growth and Development: Biology of Polyploid and Polytene Cells; Cambridge University Press: Cambridge, UK; New York, NY, USA, 1985; pp. 37–47. [Google Scholar]
- Ovrebo, J.I.; Edgar, B.A. Polyploidy in tissue homeostasis and regeneration. Development 2018, 145, dev156034. [Google Scholar] [CrossRef] [Green Version]
- Fox, D.T.; Duronio, R.J. Endoreplication and polyploidy: Insights into development and disease. Development 2013, 140, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with purpose. Genes Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef] [Green Version]
- Orr-Weaver, T.L. When bigger is better: The role of polyploidy in organogenesis. Trends Genet. 2015, 31, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Shu, Z.; Row, S.; Deng, W.M. Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef]
- Adler, C.P.; Friedburg, H. Myocardial DNA content, ploidy level and cell number in geriatric hearts: Post-mortem examinations of human myocardium in old age. J. Mol. Cell Cardiol. 1986, 18, 39–53. [Google Scholar] [CrossRef]
- Beltrami, C.A.; Di Loreto, C.; Finato, N.; Yan, S.M. DNA Content in End-Stage Heart Failure. Adv. Clin. Path 1997, 1, 59–73. [Google Scholar]
- Herget, G.W.; Neuburger, M.; Plagwitz, R.; Adler, C.P. DNA content, ploidy level and number of nuclei in the human heart after myocardial infarction. Cardiovasc. Res. 1997, 36, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Cappella, P.; Gasparri, F.; Pulici, M.; Moll, J. A novel method based on click chemistry, which overcomes limitations of cell cycle analysis by classical determination of BrdU incorporation, allowing multiplex antibody staining. Cytom. A 2008, 73, 626–636. [Google Scholar] [CrossRef]
- Leif, R.C.; Stein, J.H.; Zucker, R.M. A short history of the initial application of anti-5-BrdU to the detection and measurement of S phase. Cytom. A 2004, 58, 45–52. [Google Scholar] [CrossRef]
- Benmaamar, R.; Pagano, M. Involvement of the SCF complex in the control of Cdh1 degradation in S-phase. Cell Cycle 2005, 4, 1230–1232. [Google Scholar] [CrossRef]
- Nishitani, H.; Lygerou, Z.; Nishimoto, T. Proteolysis of DNA replication licensing factor Cdt1 in S-phase is performed independently of geminin through its N-terminal region. J. Biol. Chem. 2004, 279, 30807–30816. [Google Scholar] [CrossRef] [Green Version]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodermaier, H.C. APC/C and SCF: Controlling each other and the cell cycle. Curr. Biol. 2004, 14, R787–R796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Ayad, N.G.; Wan, Y.; Zhang, G.J.; Kirschner, M.W.; Kaelin, W.G., Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004, 428, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Zachariae, W.; Nasmyth, K. Whose end is destruction: Cell division and the anaphase-promoting complex. Genes Dev. 1999, 13, 2039–2058. [Google Scholar] [CrossRef]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar] [CrossRef]
- Abe, T.; Sakaue-Sawano, A.; Kiyonari, H.; Shioi, G.; Inoue, K.; Horiuchi, T.; Nakao, K.; Miyawaki, A.; Aizawa, S.; Fujimori, T. Visualization of cell cycle in mouse embryos with Fucci2 reporter directed by Rosa26 promoter. Development 2013, 140, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, R., Jr.; Wang, B.J.; Quijada, P.J.; Avitabile, D.; Ho, T.; Shaitrit, M.; Chavarria, M.; Firouzi, F.; Ebeid, D.; Monsanto, M.M.; et al. Cardiomyocyte cell cycle dynamics and proliferation revealed through cardiac-specific transgenesis of fluorescent ubiquitinated cell cycle indicator (FUCCI). J. Mol. Cell Cardiol. 2019, 127, 154–164. [Google Scholar] [CrossRef]
- Mort, R.L.; Ford, M.J.; Sakaue-Sawano, A.; Lindstrom, N.O.; Casadio, A.; Douglas, A.T.; Keighren, M.A.; Hohenstein, P.; Miyawaki, A.; Jackson, I.J. Fucci2a: A bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice. Cell Cycle 2014, 13, 2681–2696. [Google Scholar] [CrossRef] [Green Version]
- Basak, O.; van de Born, M.; Korving, J.; Beumer, J.; van der Elst, S.; van Es, J.H.; Clevers, H. Mapping early fate determination in Lgr5+ crypt stem cells using a novel Ki67-RFP allele. EMBO J. 2014, 33, 2057–2068. [Google Scholar] [CrossRef]
- Kretzschmar, K.; Post, Y.; Bannier-Helaouet, M.; Mattiotti, A.; Drost, J.; Basak, O.; Li, V.S.W.; van den Born, M.; Gunst, Q.D.; Versteeg, D.; et al. Profiling proliferative cells and their progeny in damaged murine hearts. Proc. Natl. Acad. Sci. USA 2018, 115, E12245–E12254. [Google Scholar] [CrossRef] [Green Version]
- Hesse, M.; Raulf, A.; Pilz, G.A.; Haberlandt, C.; Klein, A.M.; Jabs, R.; Zaehres, H.; Fugemann, C.J.; Zimmermann, K.; Trebicka, J.; et al. Direct visualization of cell division using high-resolution imaging of M-phase of the cell cycle. Nat. Commun. 2012, 3, 1076. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.; Engleka, K.A.; Liu, F.; Li, L.; Song, G.; Epstein, J.A.; Li, D. An Engineered Mouse to Identify Proliferating Cells and Their Derivatives. Front. Cell Dev. Biol. 2020, 8, 388. [Google Scholar] [CrossRef]
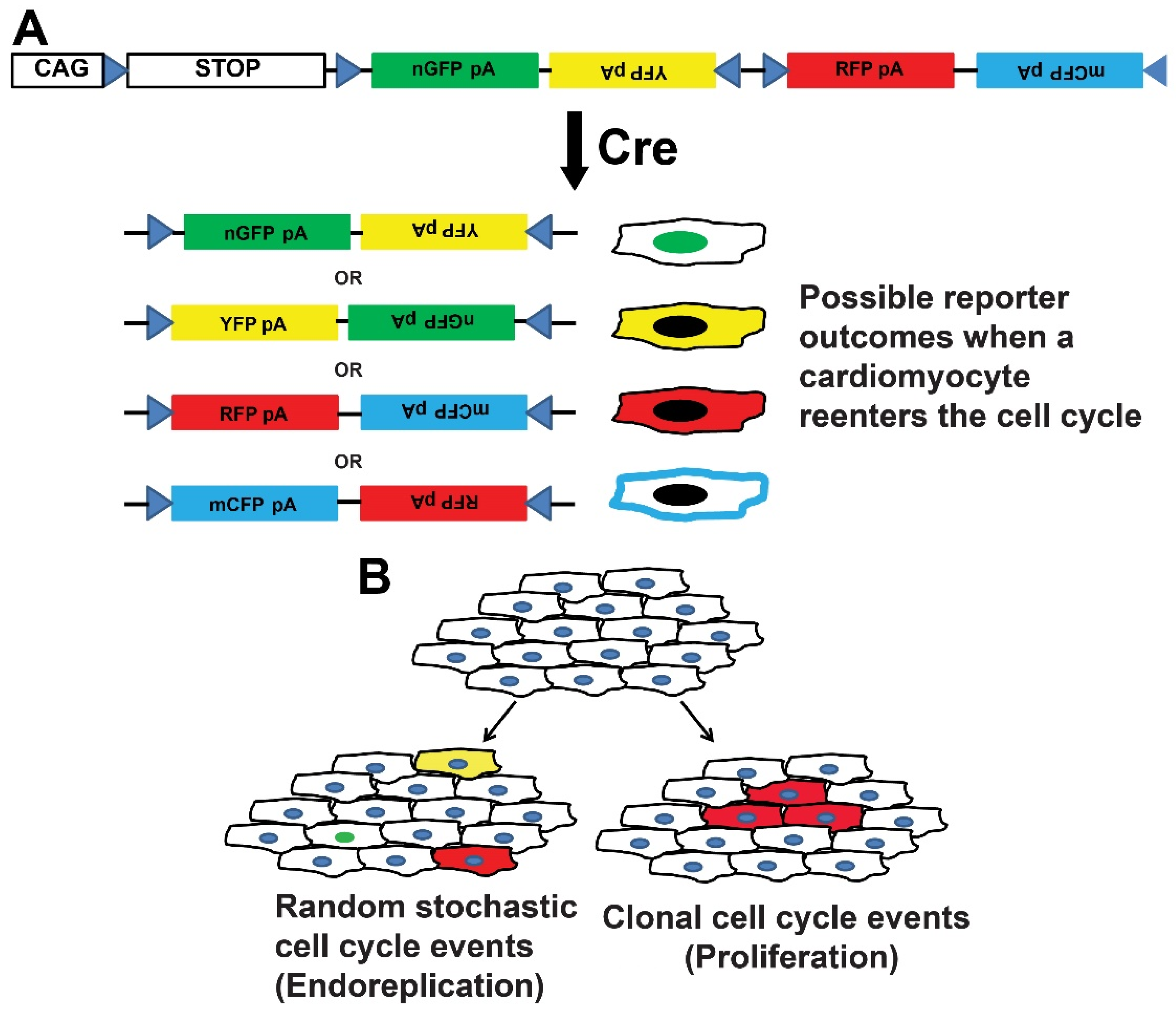
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef]
- Cai, D.; Cohen, K.B.; Luo, T.; Lichtman, J.W.; Sanes, J.R. Improved tools for the Brainbow toolbox. Nat. Methods 2013, 10, 540–547. [Google Scholar] [CrossRef]
- Lichtman, J.W.; Livet, J.; Sanes, J.R. A technicolour approach to the connectome. Nat. Rev. Neurosci. 2008, 9, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Fioret, B.A.; Heimfeld, J.D.; Paik, D.T.; Hatzopoulos, A.K. Endothelial cells contribute to generation of adult ventricular myocytes during cardiac homeostasis. Cell Rep. 2014, 8, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Gillich, A.; Zhang, F.; Farmer, C.G.; Travaglini, K.J.; Tan, S.Y.; Gu, M.; Zhou, B.; Feinstein, J.A.; Krasnow, M.A.; Metzger, R.J. Capillary cell-type specialization in the alveolus. Nature 2020, 586, 785–789. [Google Scholar] [CrossRef]
- Manavski, Y.; Lucas, T.; Glaser, S.F.; Dorsheimer, L.; Gunther, S.; Braun, T.; Rieger, M.A.; Zeiher, A.M.; Boon, R.A.; Dimmeler, S. Clonal Expansion of Endothelial Cells Contributes to Ischemia-Induced Neovascularization. Circ. Res. 2018, 122, 670–677. [Google Scholar] [CrossRef]
- Gupta, V.; Poss, K.D. Clonally dominant cardiomyocytes direct heart morphogenesis. Nature 2012, 484, 479–484. [Google Scholar] [CrossRef] [Green Version]
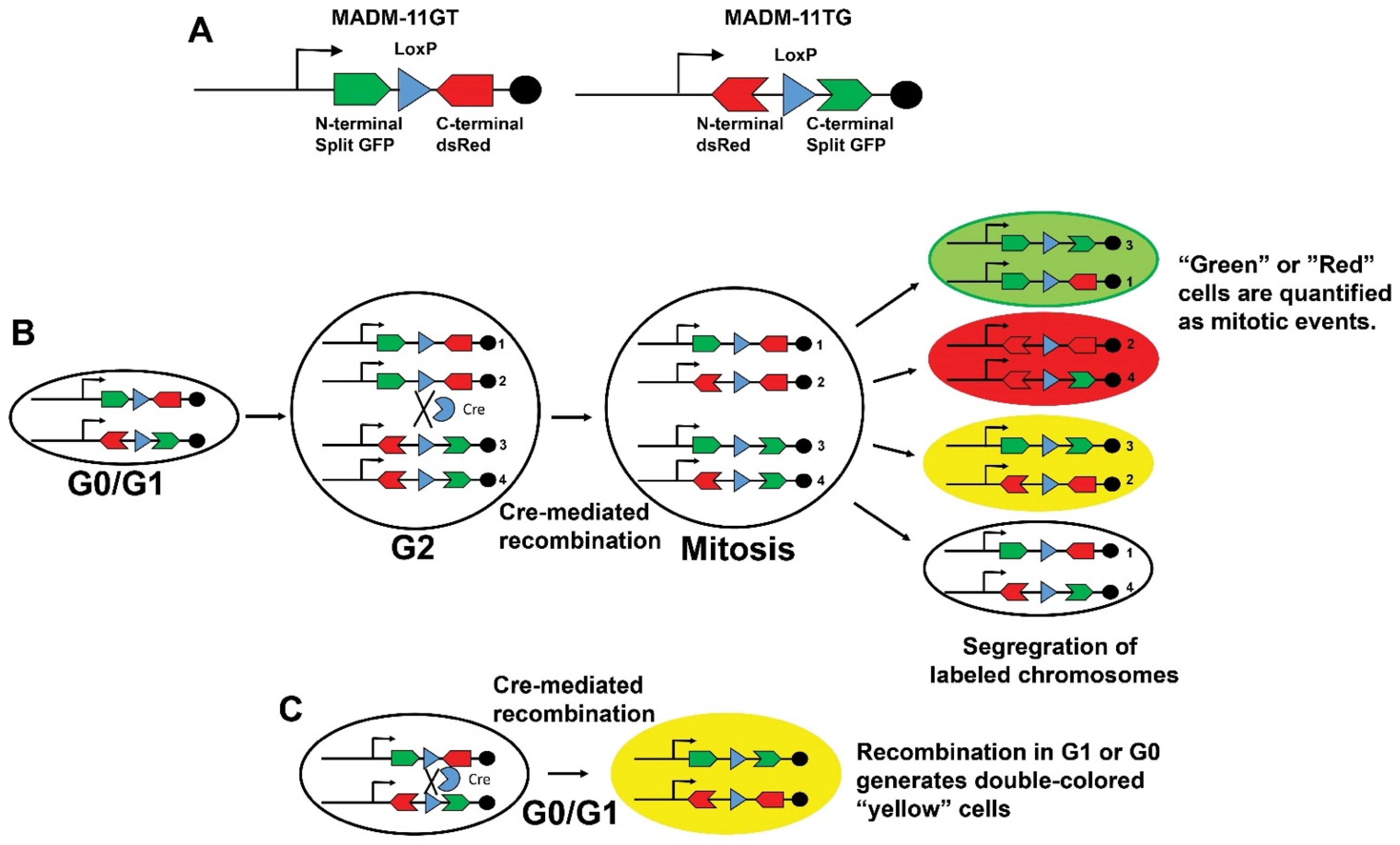
- Zong, H. Generation and applications of MADM-based mouse genetic mosaic system. Methods Mol. Biol. 2014, 1194, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Espinosa, J.S.; Su, H.H.; Muzumdar, M.D.; Luo, L. Mosaic analysis with double markers in mice. Cell 2005, 121, 479–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henner, A.; Ventura, P.B.; Jiang, Y.; Zong, H. MADM-ML, a mouse genetic mosaic system with increased clonal efficiency. PLoS ONE 2013, 8, e77672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, X.; Amberg, N.; Davaatseren, A.; Hansen, A.H.; Sonntag, J.; Andersen, L.; Bernthaler, T.; Streicher, C.; Heger, A.; Johnson, R.L.; et al. A genome-wide library of MADM mice for single-cell genetic mosaic analysis. Cell Rep. 2021, 35, 109274. [Google Scholar] [CrossRef]
- Ali, S.R.; Hippenmeyer, S.; Saadat, L.V.; Luo, L.; Weissman, I.L.; Ardehali, R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 8850–8855. [Google Scholar] [CrossRef] [Green Version]
- Magadum, A.; Singh, N.; Kurian, A.A.; Munir, I.; Mehmood, T.; Brown, K.; Sharkar, M.T.K.; Chepurko, E.; Sassi, Y.; Oh, J.G.; et al. Pkm2 Regulates Cardiomyocyte Cell Cycle and Promotes Cardiac Regeneration. Circulation 2020, 141, 1249–1265. [Google Scholar] [CrossRef]
- Nguyen, N.U.N.; Canseco, D.C.; Xiao, F.; Nakada, Y.; Li, S.; Lam, N.T.; Muralidhar, S.A.; Savla, J.J.; Hill, J.A.; Le, V.; et al. A calcineurin-Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nature 2020, 582, 271–276. [Google Scholar] [CrossRef]
- Mohamed, T.M.A.; Ang, Y.S.; Radzinsky, E.; Zhou, P.; Huang, Y.; Elfenbein, A.; Foley, A.; Magnitsky, S.; Srivastava, D. Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell 2018, 173, 104–116.e112. [Google Scholar] [CrossRef] [Green Version]
- Anastassiadis, K.; Fu, J.; Patsch, C.; Hu, S.; Weidlich, S.; Duerschke, K.; Buchholz, F.; Edenhofer, F.; Stewart, A.F. Dre recombinase, like Cre, is a highly efficient site-specific recombinase in E. coli, mammalian cells and mice. Dis. Model. Mech. 2009, 2, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Zhang, Z.; He, L.; Zhu, H.; Li, Y.; Pu, W.; Han, M.; Zhao, H.; Liu, K.; Li, Y.; et al. A suite of new Dre recombinase drivers markedly expands the ability to perform intersectional genetic targeting. Cell Stem Cell 2021, 28, 1160–1176.e7. [Google Scholar] [CrossRef]
- Sauer, B.; McDermott, J. DNA recombination with a heterospecific Cre homolog identified from comparison of the pac-c1 regions of P1-related phages. Nucleic Acids Res. 2004, 32, 6086–6095. [Google Scholar] [CrossRef]
- Kishimoto, K.; Nakayama, M.; Kinoshita, M. In vivo recombination efficiency of two site-specific recombination systems, VCre/VloxP and SCre/SloxP, in medaka (Oryzias latipes). Dev. Growth Differ. 2016, 58, 516–521. [Google Scholar] [CrossRef]
- Suzuki, E.; Nakayama, M. VCre/VloxP and SCre/SloxP: New site-specific recombination systems for genome engineering. Nucleic Acids Res. 2011, 39, e49. [Google Scholar] [CrossRef]
- Gates, C.A.; Cox, M.M. FLP recombinase is an enzyme. Proc. Natl. Acad. Sci. USA 1988, 85, 4628–4632. [Google Scholar] [CrossRef] [Green Version]
- Senecoff, J.F.; Bruckner, R.C.; Meyer-Leon, L.; Gates, C.A.; Wood, E.; Umlauf, S.W.; Attwood, J.M.; Cox, M.M. Site-specific recombination promoted in vitro by the FLP protein of the yeast two-micron plasmid. Basic Life Sci. 1986, 40, 397–405. [Google Scholar] [CrossRef]
- He, L.; Li, Y.; Li, Y.; Pu, W.; Huang, X.; Tian, X.; Wang, Y.; Zhang, H.; Liu, Q.; Zhang, L.; et al. Enhancing the precision of genetic lineage tracing using dual recombinases. Nat. Med. 2017, 23, 1488–1498. [Google Scholar] [CrossRef]
- He, L.; Huang, X.; Kanisicak, O.; Li, Y.; Wang, Y.; Li, Y.; Pu, W.; Liu, Q.; Zhang, H.; Tian, X.; et al. Preexisting endothelial cells mediate cardiac neovascularization after injury. J. Clin. Investig. 2017, 127, 2968–2981. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, X.; He, L.; Pu, W.; Li, Y.; Liu, Q.; Li, Y.; Zhang, L.; Yu, W.; Zhao, H.; et al. Genetic tracing of hepatocytes in liver homeostasis, injury, and regeneration. J. Biol. Chem. 2017, 292, 8594–8604. [Google Scholar] [CrossRef] [Green Version]
- Sajgo, S.; Ghinia, M.G.; Shi, M.; Liu, P.; Dong, L.; Parmhans, N.; Popescu, O.; Badea, T.C. Dre-Cre sequential recombination provides new tools for retinal ganglion cell labeling and manipulation in mice. PLoS ONE 2014, 9, e91435. [Google Scholar] [CrossRef] [Green Version]
- Zambon, A.C. Use of the Ki67 promoter to label cell cycle entry in living cells. Cytom. A 2010, 77, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Plummer, N.W.; Evsyukova, I.Y.; Robertson, S.D.; de Marchena, J.; Tucker, C.J.; Jensen, P. Expanding the power of recombinase-based labeling to uncover cellular diversity. Development 2015, 142, 4385–4393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersell, K.; Choudhury, S.; Mollova, M.; Polizzotti, B.D.; Ganapathy, B.; Walsh, S.; Wadugu, B.; Arab, S.; Kuhn, B. Moderate and high amounts of tamoxifen in alphaMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis. Model. Mech. 2013, 6, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Pu, W.; Zhang, M.; Liu, X.; He, L.; Li, J.; Han, X.; Lui, K.O.; He, B.; Zhou, B. Genetic Proliferation Tracing Reveals a Rapid Cell Cycle Withdrawal in Preadolescent Cardiomyocytes. Circulation 2022, 145, 410–412. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, A.; Bradley, L.A.; Wolf, M.J. In Vivo Methods to Monitor Cardiomyocyte Proliferation. J. Cardiovasc. Dev. Dis. 2022, 9, 73. https://doi.org/10.3390/jcdd9030073
Young A, Bradley LA, Wolf MJ. In Vivo Methods to Monitor Cardiomyocyte Proliferation. Journal of Cardiovascular Development and Disease. 2022; 9(3):73. https://doi.org/10.3390/jcdd9030073
Chicago/Turabian StyleYoung, Alexander, Leigh A. Bradley, and Matthew J. Wolf. 2022. "In Vivo Methods to Monitor Cardiomyocyte Proliferation" Journal of Cardiovascular Development and Disease 9, no. 3: 73. https://doi.org/10.3390/jcdd9030073