
Inhibition of the NOTCH1 Pathway in the Stressed Heart Limits Fibrosis and Promotes Recruitment of Non-Myocyte Cells into the Cardiomyocyte Fate


Abstract
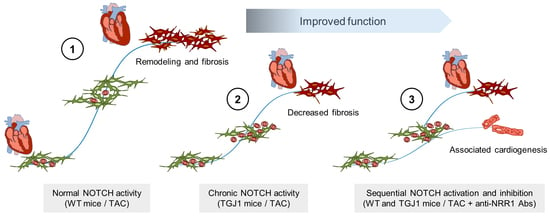
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Lineage Tracing
2.3. Surgery and Echocardiography
2.4. Antibody, BrdU and GapmeR Administration
2.5. RNA Isolation and Quantitative RT-PCR Analysis
2.6. Immunofluorescence Staining
2.7. Isolation of CMs and Non-Myocyte Cells from Adult Mouse Hearts
2.8. Histology and Fibrosis Measurements
2.9. Transfection of P19Cl6 Cells and Activation of Wisper Expression
2.10. Statistical Analysis
3. Results
3.1. The NOTCH Signaling Pathway Is Activated in Cardiac Non-Myocyte Cells Isolated from the Stressed Heart
3.2. NOTCH1 Blockade Inhibits Pressure Overload-Induced Hypertrophy and Preserves Cardiac Function
3.3. NOTCH1 Blockade Reduces Cardiac Fibrosis in the Stressed Heart
3.4. NOTCH1-Activated Cells Contribute to Non-Myocyte Cell Formation
3.5. NOTCH Blockade Stimulates CM Formation from Non-Myocyte Cells in the Stressed Heart
3.6. NOTCH-Independent Reduction in Cardiac Fibrosis Favors New CM Formation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, S.R.; Ranjbarvaziri, S.; Talkhabi, M.; Zhao, P.; Subat, A.; Hojjat, A.; Kamran, P.; Muller, A.M.; Volz, K.S.; Tang, Z.; et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 2014, 115, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamo, T.; Akazawa, H.; Komuro, I. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ. Res. 2015, 117, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senyo, S.E.; Lee, R.T.; Kuhn, B. Cardiac regeneration based on mechanisms of cardiomyocyte proliferation and differentiation. Stem Cell Res. 2014, 13, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Berlo, J.H.; Molkentin, J.D. An emerging consensus on cardiac regeneration. Nat. Med. 2014, 20, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Bersell, K.; Arab, S.; Haring, B.; Kuhn, B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009, 138, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Breckwoldt, K.; Weinberger, F.; Eschenhagen, T. Heart regeneration. Biochim. Biophys. Acta 2016, 1863, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Bolli, R.; Braun, T.; Field, L.J.; Fleischmann, B.K.; Frisen, J.; Giacca, M.; Hare, J.M.; Houser, S.; Lee, R.T.; et al. Cardiomyocyte Regeneration: A Consensus Statement. Circulation 2017, 136, 680–686. [Google Scholar] [CrossRef]
- Rockman, H.A.; Ross, R.S.; Harris, A.N.; Knowlton, K.U.; Steinhelper, M.E.; Field, L.J.; Ross, J., Jr.; Chien, K.R. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 1991, 88, 8277–8281. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- Micheletti, R.; Plaisance, I.; Abraham, B.J.; Sarre, A.; Ting, C.C.; Alexanian, M.; Maric, D.; Maison, D.; Nemir, M.; Young, R.A.; et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci. Transl. Med. 2017, 9, eaai9118. [Google Scholar] [CrossRef] [Green Version]
- Gordon, W.R.; Vardar-Ulu, D.; Histen, G.; Sanchez-Irizarry, C.; Aster, J.C.; Blacklow, S.C. Structural basis for autoinhibition of Notch. Nat. Struct. Mol. Biol. 2007, 14, 295–300. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Campa, V.M.; Gutierrez-Lanza, R.; Cerignoli, F.; Diaz-Trelles, R.; Nelson, B.; Tsuji, T.; Barcova, M.; Jiang, W.; Mercola, M. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J. Cell Biol. 2008, 183, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Collesi, C.; Zentilin, L.; Sinagra, G.; Giacca, M. Notch1 signaling stimulates proliferation of immature cardiomyocytes. J. Cell Biol. 2008, 183, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.C.; Stull, R.; Joo, D.; Cheng, X.; Keller, G. Notch signaling respecifies the hemangioblast to a cardiac fate. Nat. Biotechnol. 2008, 26, 1169–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, P.; Liu, K.; He, Q.; Han, S.; Sun, X.; Li, T.; Shen, L. Timely inhibition of Notch signaling by DAPT promotes cardiac differentiation of murine pluripotent stem cells. PLoS ONE 2014, 9, e109588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowell, S.; Benchoua, A.; Heavey, B.; Smith, A.G. Notch promotes neural lineage entry by pluripotent embryonic stem cells. PLoS Biol. 2006, 4, e121. [Google Scholar] [CrossRef] [PubMed]
- Nemir, M.; Croquelois, A.; Pedrazzini, T.; Radtke, F. Induction of cardiogenesis in embryonic stem cells via downregulation of Notch1 signaling. Circ. Res. 2006, 98, 1471–1478. [Google Scholar] [CrossRef] [Green Version]
- Tung, J.C.; Paige, S.L.; Ratner, B.D.; Murry, C.E.; Giachelli, C.M. Engineered biomaterials control differentiation and proliferation of human-embryonic-stem-cell-derived cardiomyocytes via timed Notch activation. Stem Cell Rep. 2014, 2, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Croquelois, A.; Domenighetti, A.A.; Nemir, M.; Lepore, M.; Rosenblatt-Velin, N.; Radtke, F.; Pedrazzini, T. Control of the adaptive response of the heart to stress via the Notch1 receptor pathway. J. Exp. Med. 2008, 205, 3173–3185. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.L.; Goetsch, S.C.; Gaiano, N.R.; Hill, J.A.; Olson, E.N.; Schneider, J.W. A dynamic notch injury response activates epicardium and contributes to fibrosis repair. Circ. Res. 2011, 108, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Nemir, M.; Metrich, M.; Plaisance, I.; Lepore, M.; Cruchet, S.; Berthonneche, C.; Sarre, A.; Radtke, F.; Pedrazzini, T. The Notch pathway controls fibrotic and regenerative repair in the adult heart. Eur. Heart J. 2014, 35, 2174–2185. [Google Scholar] [CrossRef] [Green Version]
- Abad, M.; Hashimoto, H.; Zhou, H.; Morales, M.G.; Chen, B.; Bassel-Duby, R.; Olson, E.N. Notch Inhibition Enhances Cardiac Reprogramming by Increasing MEF2C Transcriptional Activity. Stem Cell Rep. 2017, 8, 548–560. [Google Scholar] [CrossRef]
- Vooijs, M.; Ong, C.T.; Hadland, B.; Huppert, S.; Liu, Z.; Korving, J.; van den Born, M.; Stappenbeck, T.; Wu, Y.; Clevers, H.; et al. Mapping the consequence of Notch1 proteolysis in vivo with NIP-CRE. Development 2007, 134, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.S.; Nghiem, M.; Crackower, M.A.; Witt, S.A.; Kimball, T.R.; Tymitz, K.M.; Penninger, J.M.; Molkentin, J.D. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 2001, 89, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Metrich, M.; Bezdek Pomey, A.; Berthonneche, C.; Sarre, A.; Nemir, M.; Pedrazzini, T. Jagged1 intracellular domain-mediated inhibition of Notch1 signalling regulates cardiac homeostasis in the postnatal heart. Cardiovasc. Res. 2015, 108, 74–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqi, S.; Sussman, M.A. The heart: Mostly postmitotic or mostly premitotic? Myocyte cell cycle, senescence, and quiescence. Can. J. Cardiol. 2014, 30, 1270–1278. [Google Scholar] [CrossRef] [Green Version]
- Soonpaa, M.H.; Field, L.J. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am. J. Physiol. 1997, 272, H220–H226. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Nguyen, N.B.; Pezhouman, A.; Ardehali, R. Cardiac fibrosis: Potential therapeutic targets. Transl. Res. 2019, 209, 121–137. [Google Scholar] [CrossRef]
- Piccoli, M.T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef]
- Nistri, S.; Sassoli, C.; Bani, D. Notch Signaling in Ischemic Damage and Fibrosis: Evidence and Clues from the Heart. Front. Pharm. 2017, 8, 187. [Google Scholar] [CrossRef] [Green Version]
- Boopathy, A.V.; Martinez, M.D.; Smith, A.W.; Brown, M.E.; Garcia, A.J.; Davis, M.E. Intramyocardial Delivery of Notch Ligand-Containing Hydrogels Improves Cardiac Function and Angiogenesis Following Infarction. Tissue Eng. Part A 2015, 21, 2315–2322. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.; Sassi, Y.; Troncone, L.; Benard, L.; Ishikawa, K.; Gordon, R.E.; Lamas, S.; Laborda, J.; Hajjar, R.J.; Lebeche, D. Deletion of delta-like 1 homologue accelerates fibroblast-myofibroblast differentiation and induces myocardial fibrosis. Eur. Heart J. 2019, 40, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.H.; Dong, H.; Pan, Q.; Cao, Y.J.; Li, H.; Wang, H.C. Notch signaling may negatively regulate neonatal rat cardiac fibroblast-myofibroblast transformation. Physiol. Res. 2011, 60, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Dees, C.; Zerr, P.; Tomcik, M.; Beyer, C.; Horn, A.; Akhmetshina, A.; Palumbo, K.; Reich, N.; Zwerina, J.; Sticherling, M.; et al. Inhibition of Notch signaling prevents experimental fibrosis and induces regression of established fibrosis. Arthritis Rheum 2011, 63, 1396–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Jiang, Z.; Ruan, B.; Duan, J.; Song, P.; Liu, J.; Han, H.; Wang, L. Disruption of myofibroblastic Notch signaling attenuates liver fibrosis by modulating fibrosis progression and regression. Int. J. Biol. Sci. 2021, 17, 2135–2146. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, G.; Mo, B.; Wang, C. Artesunate ameliorates lung fibrosis via inhibiting the Notch signaling pathway. Exp. Ther. Med. 2017, 14, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Wang, W.; Cui, G.; Yan, L.; Zhang, S. Potential role of the Jagged1/Notch1 signaling pathway in the endothelial-myofibroblast transition during BLM-induced pulmonary fibrosis. J. Cell. Physiol. 2018, 233, 2451–2463. [Google Scholar] [CrossRef]
- Li, Y.; Lui, K.O.; Zhou, B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat. Rev. Cardiol. 2018, 15, 445–456. [Google Scholar] [CrossRef]
- Plaisance, I.; Perruchoud, S.; Fernandez-Tenorio, M.; Gonzales, C.; Ounzain, S.; Ruchat, P.; Nemir, M.; Niggli, E.; Pedrazzini, T. Cardiomyocyte Lineage Specification in Adult Human Cardiac Precursor Cells Via Modulation of Enhancer-Associated Long Noncoding RNA Expression. JACC Basic Transl. Sci. 2016, 1, 472–493. [Google Scholar] [CrossRef] [Green Version]
- Grego-Bessa, J.; Luna-Zurita, L.; del Monte, G.; Bolos, V.; Melgar, P.; Arandilla, A.; Garratt, A.N.; Zang, H.; Mukouyama, Y.S.; Chen, H.; et al. Notch signaling is essential for ventricular chamber development. Dev. Cell 2007, 12, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Nemir, M.; Pedrazzini, T. Functional role of Notch signaling in the developing and postnatal heart. J. Mol. Cell. Cardiol. 2008, 45, 495–504. [Google Scholar] [CrossRef]
- Miquerol, L.; Thireau, J.; Bideaux, P.; Sturny, R.; Richard, S.; Kelly, R.G. Endothelial plasticity drives arterial remodeling within the endocardium after myocardial infarction. Circ. Res. 2015, 116, 1765–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtado, M.B.; Costa, M.W.; Pranoto, E.A.; Salimova, E.; Pinto, A.R.; Lam, N.T.; Park, A.; Snider, P.; Chandran, A.; Harvey, R.P.; et al. Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ. Res. 2014, 114, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Furtado, M.B.; Nim, H.T.; Boyd, S.E.; Rosenthal, N.A. View from the heart: Cardiac fibroblasts in development, scarring and regeneration. Development 2016, 143, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemir, M.; Kay, M.; Maison, D.; Berthonneche, C.; Sarre, A.; Plaisance, I.; Pedrazzini, T. Inhibition of the NOTCH1 Pathway in the Stressed Heart Limits Fibrosis and Promotes Recruitment of Non-Myocyte Cells into the Cardiomyocyte Fate. J. Cardiovasc. Dev. Dis. 2022, 9, 111. https://doi.org/10.3390/jcdd9040111
Nemir M, Kay M, Maison D, Berthonneche C, Sarre A, Plaisance I, Pedrazzini T. Inhibition of the NOTCH1 Pathway in the Stressed Heart Limits Fibrosis and Promotes Recruitment of Non-Myocyte Cells into the Cardiomyocyte Fate. Journal of Cardiovascular Development and Disease. 2022; 9(4):111. https://doi.org/10.3390/jcdd9040111
Chicago/Turabian StyleNemir, Mohamed, Maryam Kay, Damien Maison, Corinne Berthonneche, Alexandre Sarre, Isabelle Plaisance, and Thierry Pedrazzini. 2022. "Inhibition of the NOTCH1 Pathway in the Stressed Heart Limits Fibrosis and Promotes Recruitment of Non-Myocyte Cells into the Cardiomyocyte Fate" Journal of Cardiovascular Development and Disease 9, no. 4: 111. https://doi.org/10.3390/jcdd9040111
APA StyleNemir, M., Kay, M., Maison, D., Berthonneche, C., Sarre, A., Plaisance, I., & Pedrazzini, T. (2022). Inhibition of the NOTCH1 Pathway in the Stressed Heart Limits Fibrosis and Promotes Recruitment of Non-Myocyte Cells into the Cardiomyocyte Fate. Journal of Cardiovascular Development and Disease, 9(4), 111. https://doi.org/10.3390/jcdd9040111