
HLHS: Power of the Chick Model


{kind=link}
Abstract
1. General Introduction
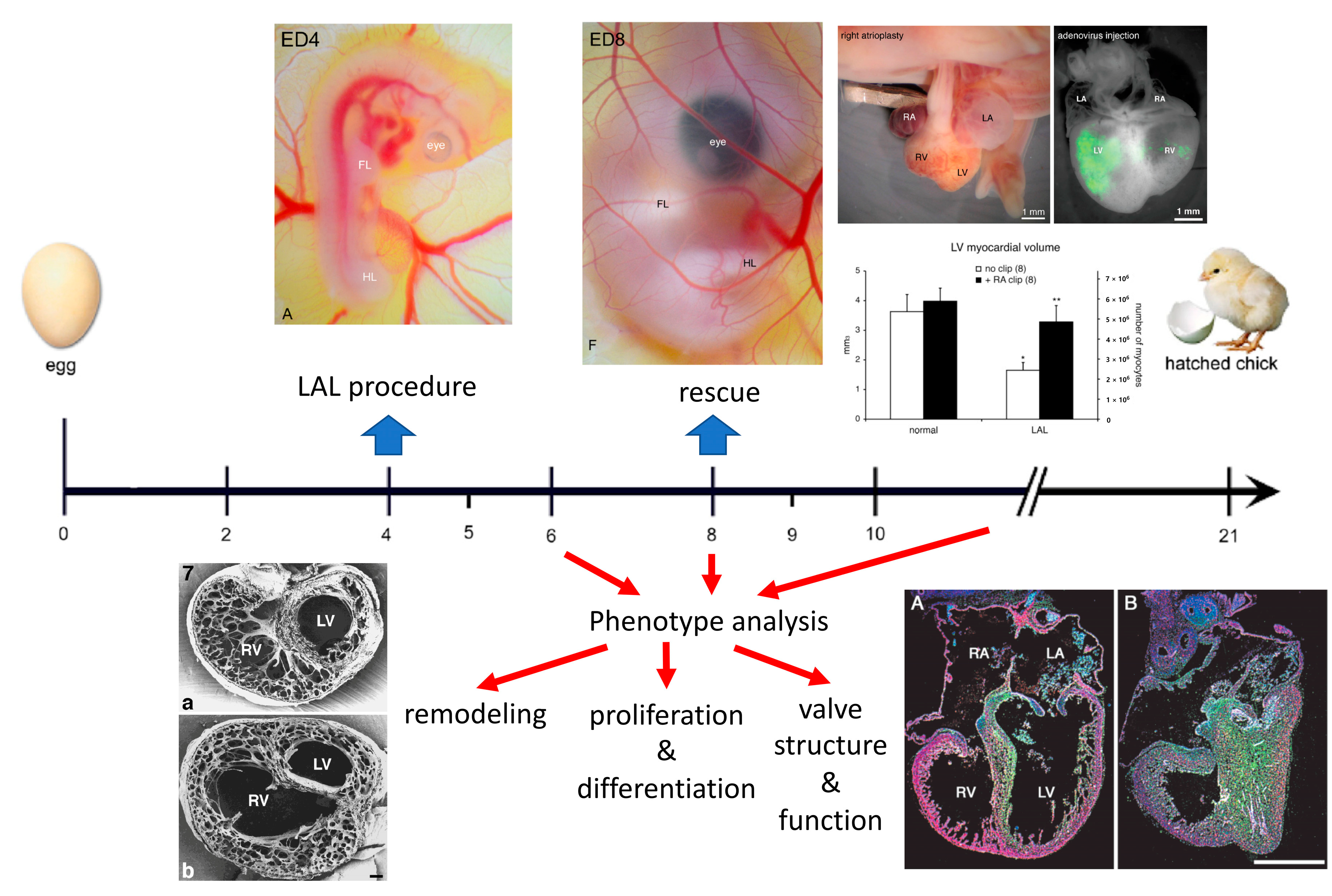
2. Surgical Creation of the Chick Model
3. Organ Level Changes Are Based upon Altered Myocyte Proliferation
4. Prenatal Phenotypic Rescue
5. Functional Adaptation to Altered Hemodynamics
6. Alterations in Electrical Conduction and Fibrosis
7. Molecular Analysis and Comparison with Human Studies
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sedmera, D.; Cook, A.C.; Shirali, G.; McQuinn, T.C. Current issues and perspectives in hypoplasia of the left heart. Cardiol. Young 2005, 15, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Fishman, N.H.; Hof, R.B.; Rudolph, A.M.; Heymann, M.A. Models of congenital heart disease in fetal lambs. Circulation 1978, 58, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Rychter, Z.; Rychterova, V.; Lemez, L. Formation of the heart loop and proliferation structure of its wall as a base for ventricular septation. Herz 1979, 4, 86–90. [Google Scholar] [PubMed]
- Rychter, Z.; Rychterova, V. Angio- and myoarchitecture of the heart wall under normal and experimentally changed morphogenesis. In Perspectives in Cardiovascular Research; Pexieder, T., Ed.; Raven Press: New York, NY, USA, 1981; Volume 5, pp. 431–452. [Google Scholar]
- Harh, J.Y.; Paul, M.H.; Gallen, W.J.; Friedberg, D.Z.; Kaplan, S. Experimental production of hypoplastic left heart syndrome in the chick embryo. Am. J. Cardiol. 1973, 31, 51–56. [Google Scholar] [CrossRef]
- Sedmera, D.; Pexieder, T.; Rychterova, V.; Hu, N.; Clark, E.B. Remodeling of chick embryonic ventricular myoarchitecture under experimentally changed loading conditions. Anat. Rec. 1999, 254, 238–252. [Google Scholar] [CrossRef]
- Rahman, A.; DeYoung, T.; Cahill, L.S.; Yee, Y.; Debebe, S.K.; Botelho, O.; Seed, M.; Chaturvedi, R.R.; Sled, J.G. A mouse model of hypoplastic left heart syndrome demonstrating left heart hypoplasia and retrograde aortic arch flow. Dis. Model Mech. 2021, 14, dmm049077. [Google Scholar] [CrossRef]
- Kockova, R.; Svatunkova, J.; Novotny, J.; Hejnova, L.; Ostadal, B.; Sedmera, D. Heart rate changes mediate the embryotoxic effect of antiarrhythmic drugs in the chick embryo. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H895–H902. [Google Scholar] [CrossRef]
- deAlmeida, A.; McQuinn, T.; Sedmera, D. Increased ventricular preload is compensated by myocyte proliferation in normal and hypoplastic fetal chick left ventricle. Circ. Res. 2007, 100, 1363–1370. [Google Scholar] [CrossRef]
- Sedmera, D.; Hu, N.; Weiss, K.M.; Keller, B.B.; Denslow, S.; Thompson, R.P. Cellular changes in experimental left heart hypoplasia. Anat. Rec. 2002, 267, 137–145. [Google Scholar] [CrossRef]
- Sedmera, D.; Pexieder, T.; Vuillemin, M.; Thompson, R.P.; Anderson, R.H. Developmental patterning of the myocardium. Anat. Rec. 2000, 258, 319–337. [Google Scholar] [CrossRef]
- de Almeida, A.; Sedmera, D. Fibroblast Growth Factor-2 regulates proliferation of cardiac myocytes in normal and hypoplastic left ventricles in the developing chick. Cardiol. Young 2009, 19, 159–169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McQuinn, T.C.; Bratoeva, M.; Dealmeida, A.; Remond, M.; Thompson, R.P.; Sedmera, D. High-frequency ultrasonographic imaging of avian cardiovascular development. Dev. Dyn. 2007, 236, 3503–3513. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.C.; van der Velde, M.E.; Tworetzky, W.; Gomez, C.A.; Wilkins-Haug, L.; Benson, C.B.; Jennings, R.W.; Lock, J.E. Creation of an atrial septal defect in utero for fetuses with hypoplastic left heart syndrome and intact or highly restrictive atrial septum. Circulation 2004, 110, 253–258. [Google Scholar] [CrossRef]
- Tworetzky, W.; Wilkins-Haug, L.; Jennings, R.W.; van der Velde, M.E.; Marshall, A.C.; Marx, G.R.; Colan, S.D.; Benson, C.B.; Lock, J.E.; Perry, S.B. Balloon dilation of severe aortic stenosis in the fetus: Potential for prevention of hypoplastic left heart syndrome: Candidate selection, technique, and results of successful intervention. Circulation 2004, 110, 2125–2131. [Google Scholar] [CrossRef]
- Marshall, A.C.; Tworetzky, W.; Bergersen, L.; McElhinney, D.B.; Benson, C.B.; Jennings, R.W.; Wilkins-Haug, L.E.; Marx, G.R.; Lock, J.E. Aortic valvuloplasty in the fetus: Technical characteristics of successful balloon dilation. J. Pediatr. 2005, 147, 535–539. [Google Scholar] [CrossRef]
- Makikallio, K.; McElhinney, D.B.; Levine, J.C.; Marx, G.R.; Colan, S.D.; Marshall, A.C.; Lock, J.E.; Marcus, E.N.; Tworetzky, W. Fetal aortic valve stenosis and the evolution of hypoplastic left heart syndrome: Patient selection for fetal intervention. Circulation 2006, 113, 1401–1405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Mustin, D.; Reardon, W.; Almeida, A.D.; Mozdziak, P.; Mrug, M.; Eisenberg, L.M.; Sedmera, D. Blood-borne stem cells differentiate into vascular and cardiac lineages during normal development. Stem Cells Dev. 2006, 15, 17–28. [Google Scholar] [CrossRef]
- Chapman, S.C.; Lawson, A.; Macarthur, W.C.; Wiese, R.J.; Loechel, R.H.; Burgos-Trinidad, M.; Wakefield, J.K.; Ramabhadran, R.; Mauch, T.J.; Schoenwolf, G.C. Ubiquitous GFP expression in transgenic chickens using a lentiviral vector. Development 2005, 132, 935–940. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Gittenberger-de Groot, A.C.; Mentink, M.M.; Bokenkamp, R.; Hogers, B. Development of the cardiac coronary vascular endothelium, studied with antiendothelial antibodies, in chicken-quail chimeras. Circ. Res. 1993, 73, 559–568. [Google Scholar] [CrossRef]
- Tobita, K.; Keller, B.B. Right and left ventricular wall deformation patterns in normal and left heart hypoplasia chick embryos. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H959–H969. [Google Scholar] [CrossRef]
- Schroder, E.A.; Tobita, K.; Tinney, J.P.; Foldes, J.K.; Keller, B.B. Microtubule involvement in the adaptation to altered mechanical load in developing chick myocardium. Circ. Res. 2002, 91, 353–359. [Google Scholar] [CrossRef]
- Tobita, K.; Schroder, E.A.; Tinney, J.P.; Garrison, J.B.; Keller, B.B. Regional passive ventricular stress-strain relations during development of altered loads in chick embryo. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2386–H2396. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Christensen, D.A.; Agrawal, A.K.; Beaumont, C.; Clark, E.B.; Hawkins, J.A. Dependence of aortic arch morphogenesis on intracardiac blood flow in the left atrial ligated chick embryo. Anat. Rec. 2009, 292, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.; Chan, W.X.; Yap, C.H. Fluid mechanics of the left atrial ligation chick embryonic model of hypoplastic left heart syndrome. Biomech. Model Mechanobiol. 2021, 20, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Reckova, M.; Rosengarten, C.; deAlmeida, A.; Stanley, C.P.; Wessels, A.; Gourdie, R.G.; Thompson, R.P.; Sedmera, D. Hemodynamics is a key epigenetic factor in development of the cardiac conduction system. Circ. Res. 2003, 93, 77–85. [Google Scholar] [CrossRef]
- Hall, C.E.; Hurtado, R.; Hewett, K.W.; Shulimovich, M.; Poma, C.P.; Reckova, M.; Justus, C.; Pennisi, D.J.; Tobita, K.; Sedmera, D.; et al. Hemodynamic-dependent patterning of endothelin converting enzyme 1 expression and differentiation of impulse-conducting Purkinje fibers in the embryonic heart. Development 2004, 131, 581–592. [Google Scholar] [CrossRef]
- Pesevski, Z.; Kvasilova, A.; Stopkova, T.; Nanka, O.; Drobna Krejci, E.; Buffinton, C.; Kockova, R.; Eckhardt, A.; Sedmera, D. Endocardial Fibroelastosis is Secondary to Hemodynamic Alterations in the Chick Embryonic Model of Hypoplastic Left Heart Syndrome. Dev. Dyn. 2018, 247, 509–520. [Google Scholar] [CrossRef]
- Krejci, E.; Pesevski, Z.; DeAlmeida, A.C.; Mrug, M.; Fresco, V.M.; Argraves, W.S.; Barth, J.L.; Cui, X.; Sedmera, D. Microarray analysis of normal and abnormal chick ventricular myocardial development. Physiol. Res. 2012, 61 (Suppl. 1), S137–S144. [Google Scholar] [CrossRef]
- Krane, M.; Dressen, M.; Santamaria, G.; My, I.; Schneider, C.M.; Dorn, T.; Laue, S.; Mastantuono, E.; Berutti, R.; Rawat, H.; et al. Sequential Defects in Cardiac Lineage Commitment and Maturation Cause Hypoplastic Left Heart Syndrome. Circulation 2021, 144, 1409–1428. [Google Scholar] [CrossRef]
- Antin, P.B.; Fallon, J.F.; Schoenwolf, G.C. The chick embryo rules (still)! Dev. Dyn. 2004, 229, 413. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedmera, D. HLHS: Power of the Chick Model. J. Cardiovasc. Dev. Dis. 2022, 9, 113. https://doi.org/10.3390/jcdd9040113
Sedmera D. HLHS: Power of the Chick Model. Journal of Cardiovascular Development and Disease. 2022; 9(4):113. https://doi.org/10.3390/jcdd9040113
Chicago/Turabian StyleSedmera, David. 2022. "HLHS: Power of the Chick Model" Journal of Cardiovascular Development and Disease 9, no. 4: 113. https://doi.org/10.3390/jcdd9040113
APA StyleSedmera, D. (2022). HLHS: Power of the Chick Model. Journal of Cardiovascular Development and Disease, 9(4), 113. https://doi.org/10.3390/jcdd9040113