
Significance of α-Myosin Heavy Chain (MYH6) Variants in Hypoplastic Left Heart Syndrome and Related Cardiovascular Diseases

 ,
,
Abstract
:1. Background
2. Genetic Studies
2.1. Known MYH6 Variant Disease Associations
2.2. Clinical Interpretation of Genetic Studies
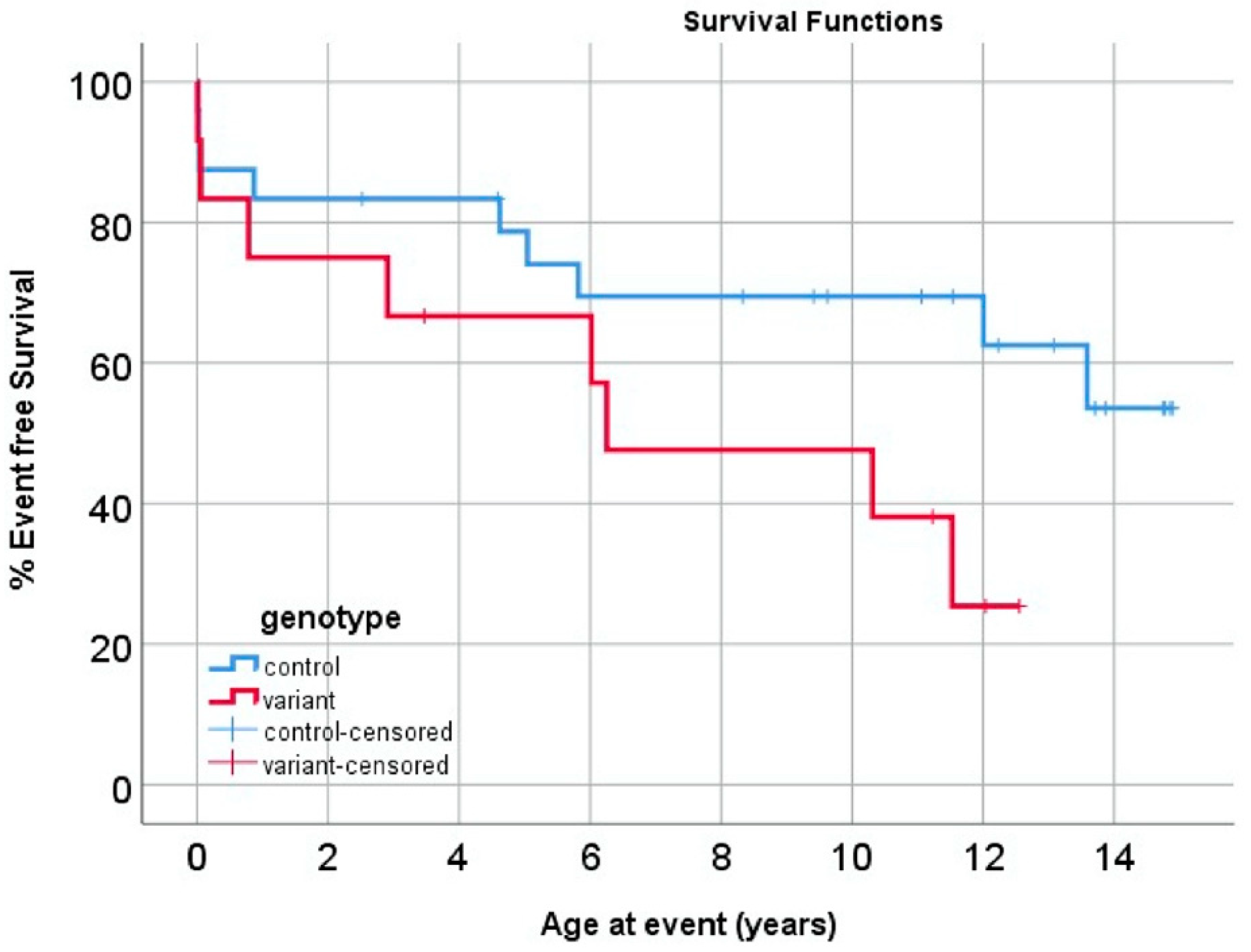
2.3. Impact of MYH6 Variants on Outcomes in HLHS
3. Mechanisms of MYH6 Variant Pathology
3.1. Importance of Mechanistic Studies
3.2. In Vitro Mechanistic Studies
3.3. In Vivo Mechanistic Studies
3.4. Structural Considerations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tchervenkov, C.I.; Jacobs, M.L.; Tahta, S.A. Congenital Heart Surgery Nomenclature and Database Project: Hypoplastic left heart syndrome. Ann. Thorac. Surg. 2000, 69, S170–S179. [Google Scholar] [CrossRef]
- McBride, K.L.; Pignatelli, R.; Lewin, M.; Ho, T.; Fernbach, S.; Menesses, A.; Lam, W.; Leal, S.M.; Kaplan, N.; Schliekelman, P.; et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: Segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. A 2005, 134A, 180–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinton, R.B., Jr.; Martin, L.J.; Tabangin, M.E.; Mazwi, M.L.; Cripe, L.H.; Benson, D.W. Hypoplastic left heart syndrome is heritable. J. Am. Coll. Cardiol. 2007, 50, 1590–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara, D.A.; Ethen, M.K.; Canfield, M.A.; Nembhard, W.N.; Morris, S.A. A population-based analysis of mortality in patients with Turner syndrome and hypoplastic left heart syndrome using the Texas Birth Defects Registry. Congenit. Heart Dis. 2017, 12, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Grossfeld, P.D.; Mattina, T.; Lai, Z.; Favier, R.; Jones, K.L.; Cotter, F.; Jones, C. The 11q terminal deletion disorder: A prospective study of 110 cases. Am. J. Med. Genet. A 2004, 129A, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yagi, H.; Saeed, S.; Bais, A.S.; Gabriel, G.C.; Chen, Z.; Peterson, K.A.; Li, Y.; Schwartz, M.C.; Reynolds, W.T.; et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017, 49, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Liu, X.; Gabriel, G.C.; Wu, Y.; Peterson, K.; Murray, S.A.; Aronow, B.J.; Martin, L.J.; Benson, D.W.; Lo, C.W. The Genetic Landscape of Hypoplastic Left Heart Syndrome. Pediatr. Cardiol. 2018, 39, 1069–1081. [Google Scholar] [CrossRef]
- Tomita-Mitchell, A.; Stamm, K.D.; Mahnke, D.K.; Kim, M.S.; Hidestrand, P.M.; Liang, H.L.; Goetsch, M.A.; Hidestrand, M.; Simpson, P.; Pelech, A.N.; et al. Impact of MYH6 variants in hypoplastic left heart syndrome. Physiol. Genom. 2016, 48, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Theis, J.L.; Zimmermann, M.T.; Evans, J.M.; Eckloff, B.W.; Wieben, E.D.; Qureshi, M.Y.; O’Leary, P.W.; Olson, T.M. Recessive MYH6 Mutations in Hypoplastic Left Heart With Reduced Ejection Fraction. Circ. Cardiovasc. Genet. 2015, 8, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Xu, Y.; Yu, M.; Lee, D.; Alharti, S.; Hellen, N.; Ahmad Shaik, N.; Banaganapalli, B.; Sheikh Ali Mohamoud, H.; Elango, R.; et al. Induced pluripotent stem cell modelling of HLHS underlines the contribution of dysfunctional NOTCH signalling to impaired cardiogenesis. Hum. Mol. Genet. 2017, 26, 3031–3045. [Google Scholar] [CrossRef]
- Elliott, D.A.; Kirk, E.P.; Yeoh, T.; Chandar, S.; McKenzie, F.; Taylor, P.; Grossfeld, P.; Fatkin, D.; Jones, O.; Hayes, P.; et al. Cardiac homeobox gene NKX2-5mutations and congenital heart disease. J. Am. Coll. Cardiol. 2003, 41, 2072–2076. [Google Scholar] [CrossRef] [Green Version]
- McBride, K.L.; Zender, G.A.; Fitzgerald-Butt, S.M.; Seagraves, N.J.; Fernbach, S.D.; Zapata, G.; Lewin, M.; Towbin, J.A.; Belmont, J.W. Association of common variants in ERBB4 with congenital left ventricular outflow tract obstruction defects. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 162–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reamon-Buettner, S.M.; Ciribilli, Y.; Inga, A.; Borlak, J. A loss-of-function mutation in the binding domain of HAND1 predicts hypoplasia of the human hearts. Hum. Mol. Genet. 2008, 17, 1397–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, C.; Martinez, A.M.; Zuppan, C.W.; Shah, M.M.; Bailey, L.L.; Fletcher, W.H. Identification of connexin43 (alpha1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (DGGE). Mutat. Res. 2001, 479, 173–186. [Google Scholar] [CrossRef]
- Berdougo, E.; Coleman, H.; Lee, D.H.; Stainier, D.Y.; Yelon, D. Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development 2003, 130, 6121–6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auman, H.J.; Coleman, H.; Riley, H.E.; Olale, F.; Tsai, H.J.; Yelon, D. Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol. 2007, 5, e53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Daya, A.; Sater, A.K.; Wells, D.E.; Mohun, T.J.; Zimmerman, L.B. Absence of heartbeat in the Xenopus tropicalis mutation muzak is caused by a nonsense mutation in cardiac myosin myh6. Dev. Biol. 2009, 336, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Reiser, P.J.; Portman, M.A.; Ning, X.H.; Moravec, C.S. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1814–H1820. [Google Scholar] [CrossRef] [Green Version]
- Wessels, A.; Vermeulen, J.L.M.; Virágh, S.Z.; Moorman, A.F.M. The ontogenesis of myosin heavy chain isoforms in the developing human heart. Ann. N. Y. Acad. Sci. 1990, 588, 461–464. [Google Scholar] [CrossRef]
- Wessels, A.; Vermeulen, J.L.M.; Virágh, S.Z.; Kálmán, F.; Lamers, W.H.; Moorman, A.F.M. Spatial distribution of “tissue-specific” antigens in the developing human heart and skeletal muscle. II. An immunohistochemical analysis of myosin heavy chain isoform expression patterns in the embryonic heart. Anat. Rec. 1991, 229, 355–368. [Google Scholar] [CrossRef]
- Solomon, S.D.; Geisterfer-Lowrance, A.; Vosberg, H.P.; Gudrun, H.; Jarcho, J.A.; Morton, C.C.; McBride, W.O.; Mitchell, A.L.; Bale, A.E.; McKenna, W.; et al. A locus for familial hypertrophic cardiomyopathy is closely linked to the cardiac myosin heavy chain genes, CRI-L436, and CRI-L329 on chromosome 14 at q11-q12. Am. J. Hum. Genet. 1990, 47, 389–394. [Google Scholar] [PubMed]
- Tanigawa, G.; Jarcho, J.A.; Kass, S.; Solomon, S.D.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. A molecular basis for familial hypertrophic cardiomyopathy: An alpha/beta cardiac myosin heavy chain hybrid gene. Cell Press 1990, 62, 991–998. [Google Scholar] [CrossRef]
- Pulignani, S.; Vecoli, C.; Borghini, A.; Foffa, I.; Ait-Ali, L.; Andreassi, M.G. Targeted Next-Generation Sequencing in Patients with Non-syndromic Congenital Heart Disease. Pediatr. Cardiol. 2018, 39, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Priest, J.R.; Osoegawa, K.; Mohammed, N.; Nanda, V.; Kundu, R.; Schultz, K.; Lammer, E.J.; Girirajan, S.; Scheetz, T.; Waggott, D.; et al. De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet. 2016, 12, e1005963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Louw, J.J.; Breckpot, J.; Callewaert, B.; Barrea, C.; Sznajer, Y.; Gewillig, M.; Souche, E.; Dehaspe, L.; Vermeesch, J.R.; et al. The diagnostic value of next generation sequencing in familial nonsyndromic congenital heart defects. Am. J. Med. Genet. A 2015, 167A, 1822–1829. [Google Scholar] [CrossRef]
- Granados-Riveron, J.T.; Ghosh, T.K.; Pope, M.; Bu’Lock, F.; Thornborough, C.; Eason, J.; Kirk, E.P.; Fatkin, D.; Feneley, M.P.; Harvey, R.P.; et al. Alpha-cardiac myosin heavy chain (MYH6) mutations affecting myofibril formation are associated with congenital heart defects. Hum. Mol. Genet. 2010, 19, 4007–4016. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Razmara, E.; Garshasbi, M. Whole-exome sequencing identifies R1279X of MYH6 gene to be associated with congenital heart disease. BMC Cardiovasc. Disord. 2018, 18, 137. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Li, Y.; Lei, D.; Li, L.; Hou, Z.L.; Han, S.; Meng, M.; Shi, J.; Zhang, Y.; et al. Novel Genetic Variants of Sporadic Atrial Septal Defect (ASD) in a Chinese Population Identified by Whole-Exome Sequencing (WES). Med. Sci. Monit. 2018, 24, 1340–1358. [Google Scholar] [CrossRef] [Green Version]
- Posch, M.G.; Waldmuller, S.; Muller, M.; Scheffold, T.; Fournier, D.; Andrade-Navarro, M.A.; De Geeter, B.; Guillaumont, S.; Dauphin, C.; Yousseff, D.; et al. Cardiac alpha-myosin (MYH6) is the predominant sarcomeric disease gene for familial atrial septal defects. PLoS ONE 2011, 6, e28872. [Google Scholar] [CrossRef]
- Ching, Y.H.; Ghosh, T.K.; Cross, S.J.; Packham, E.A.; Honeyman, L.; Loughna, S.; Robinson, T.E.; Dearlove, A.M.; Ribas, G.; Bonser, A.J.; et al. Mutation in myosin heavy chain 6 causes atrial septal defect. Nat. Genet. 2005, 37, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Bowles, N.E.; Jou, C.J.; Arrington, C.B.; Kennedy, B.J.; Earl, A.; Matsunami, N.; Meyers, L.L.; Etheridge, S.P.; Saarel, E.V.; Bleyl, S.B.; et al. Exome analysis of a family with Wolff-Parkinson-White syndrome identifies a novel disease locus. Am. J. Med. Genet. A 2015, 167A, 2975–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, L.; Ingles, J.; Turner, C.; Kilborn, M.; Bagnall, R.D.; Semsarian, C. Exome sequencing identifies a novel mutation in the MYH6 gene in a family with early-onset sinus node dysfunction, ventricular arrhythmias, and cardiac arrest. HeartRhythm Case Rep. 2015, 1, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalazan, B.; Mol, D.; Darbar, F.A.; Ornelas-Loredo, A.; Al-Azzam, B.; Chen, Y.; Tofovic, D.; Sridhar, A.; Alzahrani, Z.; Ellinor, P.; et al. Association of Rare Genetic Variants and Early-Onset Atrial Fibrillation in Ethnic Minority Individuals. JAMA Cardiol. 2021, 6, 811–819. [Google Scholar] [CrossRef]
- Holm, H.; Gudbjartsson, D.F.; Sulem, P.; Masson, G.; Helgadottir, H.T.; Zanon, C.; Magnusson, O.T.; Helgason, A.; Saemundsdottir, J.; Gylfason, A.; et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat. Genet. 2011, 43, 316–320. [Google Scholar] [CrossRef]
- Hata, Y.; Yoshida, K.; Kinoshita, K.; Nishida, N. Epilepsy-related sudden unexpected death: Targeted molecular analysis of inherited heart disease genes using next-generation DNA sequencing. Brain Pathol. 2017, 27, 292–304. [Google Scholar] [CrossRef]
- Scheiper, S.; Ramos-Luis, E.; Blanco-Verea, A.; Niess, C.; Beckmann, B.M.; Schmidt, U.; Kettner, M.; Geisen, C.; Verhoff, M.A.; Brion, M.; et al. Sudden unexpected death in the young—Value of massive parallel sequencing in postmortem genetic analyses. Forensic. Sci. Int. 2018, 293, 70–76. [Google Scholar] [CrossRef]
- Nielsen, J.B.; Thorolfsdottir, R.B.; Fritsche, L.G.; Zhou, W.; Skov, M.W.; Graham, S.E.; Herron, T.J.; McCarthy, S.; Schmidt, E.M.; Sveinbjornsson, G.; et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet. 2018, 50, 1234–1239. [Google Scholar] [CrossRef]
- Ishikawa, T.; Jou, C.J.; Nogami, A.; Kowase, S.; Arrington, C.B.; Barnett, S.M.; Harrell, D.T.; Arimura, T.; Tsuji, Y.; Kimura, A.; et al. Novel mutation in the alpha-myosin heavy chain gene is associated with sick sinus syndrome. Circ. Arrhythmia Electrophysiol. 2015, 8, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Thorolfsdottir, R.B.; Sveinbjornsson, G.; Aegisdottir, H.M.; Benonisdottir, S.; Stefansdottir, L.; Ivarsdottir, E.V.; Halldorsson, G.H.; Sigurdsson, J.K.; Torp-Pedersen, C.; Weeke, P.E.; et al. Genetic insight into sick sinus syndrome. Eur. Heart J. 2021, 42, 1959–1971. [Google Scholar] [CrossRef]
- Guelly, C.; Abilova, Z.; Nuralinov, O.; Panzitt, K.; Akhmetova, A.; Rakhimova, S.; Kozhamkulov, U.; Kairov, U.; Molkenov, A.; Seisenova, A.; et al. Patients with coronary heart disease, dilated cardiomyopathy and idiopathic ventricular tachycardia share overlapping patterns of pathogenic variation in cardiac risk genes. PeerJ 2021, 9, e10711. [Google Scholar] [CrossRef] [PubMed]
- Niimura, H.; Patton, K.K.; McKenna, W.J.; Soults, J.; Maron, B.J.; Seidman, J.G.; Seidman, C.E. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 2002, 105, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Bozzao, C.; Pennacchini, E.; Pagannone, E.; Musumeci, B.M.; Piane, M.; Germani, A.; Savio, C.; Francia, P.; Volpe, M.; et al. A Next-Generation Sequencing Approach to Identify Gene Mutations in Early- and Late-Onset Hypertrophic Cardiomyopathy Patients of an Italian Cohort. Int. J. Mol. Sci. 2016, 17, 1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, Y.; Ichimata, S.; Yamaguchi, Y.; Hirono, K.; Oku, Y.; Ichida, F.; Nishida, N. Clinicopathological and Genetic Profiles of Cases with Myocytes Disarray-Investigation for Establishing the Autopsy Diagnostic Criteria for Hypertrophic Cardiomyopathy. J. Clin. Med. 2019, 8, 463. [Google Scholar] [CrossRef] [Green Version]
- Castellana, S.; Mastroianno, S.; Palumbo, P.; Palumbo, O.; Biagini, T.; Leone, M.P.; De Luca, G.; Potenza, D.R.; Amico, C.M.; Mazza, T.; et al. Sudden death in mild hypertrophic cardiomyopathy with compound DSG2/DSC2/MYH6 mutations: Revisiting phenotype after genetic assessment in a master runner athlete. J. Electrocardiol. 2019, 53, 95–99. [Google Scholar] [CrossRef]
- Singer, E.S.; Ross, S.B.; Skinner, J.R.; Weintraub, R.G.; Ingles, J.; Semsarian, C.; Bagnall, R.D. Characterization of clinically relevant copy-number variants from exomes of patients with inherited heart disease and unexplained sudden cardiac death. Genet. Med. 2021, 23, 86–93. [Google Scholar] [CrossRef]
- Carniel, E.; Taylor, M.R.; Sinagra, G.; Di Lenarda, A.; Ku, L.; Fain, P.R.; Boucek, M.M.; Cavanaugh, J.; Miocic, S.; Slavov, D.; et al. Alpha-myosin heavy chain: A sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 2005, 112, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Norton, N.; Morales, A.; Li, D.; Siegfried, J.D.; Gonzalez-Quintana, J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Rampersaud, E.; Siegfried, J.D.; Norton, N.; Li, D.; Martin, E.; Hershberger, R.E. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog. Pediatr. Cardiol. 2011, 31, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Merlo, M.; Sinagra, G.; Carniel, E.; Slavov, D.; Zhu, X.; Barbati, G.; Spezzacatene, A.; Ramani, F.; Salcedo, E.; Di Lenarda, A.; et al. Poor prognosis of rare sarcomeric gene variants in patients with dilated cardiomyopathy. Clin. Transl. Sci. 2013, 6, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Feng, Y.; Zhang, Y.M.; Ding, X.X.; Song, Y.Z.; Zhang, A.M.; Liu, L.; Zhang, H.; Ding, J.H.; Xia, X.S. Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int. J. Mol. Med. 2015, 36, 1479–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Ma, Y.; Zhang, Z.; Xian, J.; Geng, X.; Wang, F.; Huang, J.; Yang, Z.; Luo, Y.; Lin, Y. Young and early-onset dilated cardiomyopathy with malignant ventricular arrhythmia and sudden cardiac death induced by the heterozygous LDB3, MYH6, and SYNE1 missense mutations. Ann. Noninvasive Electrocardiol. 2021, 26, e12840. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.; Painter, T.; Li, R.; Siegfried, J.D.; Li, D.; Norton, N.; Hershberger, R.E. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation 2010, 121, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Rao, M.; Guo, G.; Chen, X.; Chen, L.; Song, J. Sarcomere variants in arrhythmogenic cardiomyopathy: Pathogenic factor or bystander? Gene 2019, 687, 82–89. [Google Scholar] [CrossRef]
- Vershinina, T.; Fomicheva, Y.; Muravyev, A.; Jorholt, J.; Kozyreva, A.; Kiselev, A.; Gordeev, M.; Vasichkina, E.; Segrushichev, A.; Pervunina, T.; et al. Genetic Spectrum of Left Ventricular Non-Compaction in Paediatric Patients. Cardiology 2020, 145, 746–756. [Google Scholar] [CrossRef]
- Armes, J.E.; Squires, L.; Lourie, R.; Williams, M.; Gallagher, R.; Price, G.; Stubbs, A.; Swagemakers, S.M.; van der Spek, P.J.; Harraway, J.; et al. Isolated Ventricular Noncompaction Cardiomyopathy Presenting as Fetal Hydrops at 24 Weeks Gestation. Pediatr. Dev. Pathol. 2017, 20, 245–250. [Google Scholar] [CrossRef]
- van Wijngaarden, A.L.; Hiemstra, Y.L.; Koopmann, T.T.; Ruivenkamp, C.A.L.; Aten, E.; Schalij, M.J.; Bax, J.J.; Delgado, V.; Barge-Schaapveld, D.; Ajmone Marsan, N. Identification of known and unknown genes associated with mitral valve prolapse using an exome slice methodology. J. Med. Genet. 2020, 57, 843–850. [Google Scholar] [CrossRef]
- Bjornsson, T.; Thorolfsdottir, R.B.; Sveinbjornsson, G.; Sulem, P.; Norddahl, G.L.; Helgadottir, A.; Gretarsdottir, S.; Magnusdottir, A.; Danielsen, R.; Sigurdsson, E.L.; et al. A rare missense mutation in MYH6 associates with non-syndromic coarctation of the aorta. Eur. Heart J. 2018, 39, 3243–3249. [Google Scholar] [CrossRef]
- Hu, P.; Qiao, F.; Wang, Y.; Meng, L.; Ji, X.; Luo, C.; Xu, T.; Zhou, R.; Zhang, J.; Yu, B.; et al. Clinical application of targeted next-generation sequencing in fetuses with congenital heart defect. Ultrasound Obstet. Gynecol. 2018, 52, 205–211. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theis, J.L.; Hu, J.J.; Sundsbak, R.S.; Evans, J.M.; Bamlet, W.R.; Qureshi, M.Y.; O’Leary, P.W.; Olson, T.M. Genetic Association Between Hypoplastic Left Heart Syndrome and Cardiomyopathies. Circ. Genom. Precis. Med. 2021, 14, e003126. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Chen, S.; Yang, Y.; Wu, Y.; Huang, S.; Wang, Y.; Ding, H.; He, W.; Li, P.; Zhuang, J. Novel Mutation in MYH6 in 2 Unrelated Chinese Han Families With Familial Atrial Septal Defect. Circ. Genom. Precis. Med. 2019, 12, e002732. [Google Scholar] [CrossRef] [PubMed]
- Granados-Riveron, J.T.; Pope, M.; Bu’lock, F.A.; Thornborough, C.; Eason, J.; Setchfield, K.; Ketley, A.; Kirk, E.P.; Fatkin, D.; Feneley, M.P.; et al. Combined mutation screening of NKX2-5, GATA4, and TBX5 in congenital heart disease: Multiple heterozygosity and novel mutations. Congenit. Heart Dis. 2012, 7, 151–159. [Google Scholar] [CrossRef]
- Liu, W.; Wei, Z.; Zhang, Y.; Liu, Y.; Bai, R.; Ma, C.; Yang, J.; Sun, D. Identification of three novel pathogenic mutations in sarcomere genes associated with familial hypertrophic cardiomyopathy based on multi-omics study. Clin. Chim. Acta 2021, 520, 43–52. [Google Scholar] [CrossRef]
- Hayashi, K.; Teramoto, R.; Nomura, A.; Asano, Y.; Beerens, M.; Kurata, Y.; Kobayashi, I.; Fujino, N.; Furusho, H.; Sakata, K.; et al. Impact of functional studies on exome sequence variant interpretation in early-onset cardiac conduction system diseases. Cardiovasc. Res. 2020, 116, 2116–2130. [Google Scholar] [CrossRef]
- Newburger, J.W.; Sleeper, L.A.; Gaynor, J.W.; Hollenbeck-Pringle, D.; Frommelt, P.C.; Li, J.S.; Mahle, W.T.; Williams, I.A.; Atz, A.M.; Burns, K.M.; et al. Transplant-Free Survival and Interventions at 6 Years in the SVR Trial. Circulation 2018, 137, 2246–2253. [Google Scholar] [CrossRef] [Green Version]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sorensen, T.; et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. N. Engl. J. Med. 2021, 384, 105–116. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.J.V.; Ponikowski, P.; Metra, M.; Filippatos, G.S.; Ezekowitz, J.A.; Dickstein, K.; Cleland, J.G.F.; Kim, J.B.; et al. Acute Treatment With Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure: The ATOMIC-AHF Study. J. Am. Coll. Cardiol. 2016, 67, 1444–1455. [Google Scholar] [CrossRef] [Green Version]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.J.V.; Solomon, S.D.; Adams, K.F.; Cleland, J.G.F.; Ezekowitz, J.A.; Goudev, A.; Macdonald, P.; Metra, M.; et al. Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF): A phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet 2016, 388, 2895–2903. [Google Scholar] [CrossRef] [Green Version]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Huang, S.; Wu, Y.; Chen, S.; Yang, Y.; Wang, Y.; Wang, H.; Li, P.; Zhuang, J.; Xia, Y. Novel insertion mutation (Arg1822_Glu1823dup) in MYH6 coiled-coil domain causing familial atrial septal defect. Eur. J. Med. Genet. 2021, 64, 104314. [Google Scholar] [CrossRef] [PubMed]
- Klos, M.; Mundada, L.; Banerjee, I.; Morgenstern, S.; Myers, S.; Leone, M.; Kleid, M.; Herron, T.; Devaney, E. Altered myocyte contractility and calcium homeostasis in alpha-myosin heavy chain point mutations linked to familial dilated cardiomyopathy. Arch. Biochem. Biophys. 2017, 615, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Fleres, B.; Lovett, J.; Anfinson, M.; Samudrala, S.S.K.; Kelly, L.J.; Teigen, L.E.; Cavanaugh, M.; Marquez, M.; Geurts, A.M.; et al. Contractility of Induced Pluripotent Stem Cell-Cardiomyocytes With an MYH6 Head Domain Variant Associated With Hypoplastic Left Heart Syndrome. Front. Cell Dev. Biol. 2020, 8, 440. [Google Scholar] [CrossRef] [PubMed]
- Kampourakis, T.; Irving, M. Phosphorylation of myosin regulatory light chain controls myosin head conformation in cardiac muscle. J. Mol. Cell Cardiol. 2015, 85, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flashman, E.; Redwood, C.; Moolman-Smook, J.; Watkins, H. Cardiac myosin binding protein C: Its role in physiology and disease. Circ. Res. 2004, 94, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Homburger, J.R.; Green, E.M.; Caleshu, C.; Sunitha, M.S.; Taylor, R.E.; Ruppel, K.M.; Metpally, R.P.; Colan, S.D.; Michels, M.; Day, S.M.; et al. Multidimensional structure-function relationships in human beta-cardiac myosin from population-scale genetic variation. Proc. Natl. Acad. Sci. USA 2016, 113, 6701–6706. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.A.; Caleshu, C.; Morales, A.; Buchan, J.; Wolf, Z.; Harrison, S.M.; Cook, S.; Dillon, M.W.; Garcia, J.; Haverfield, E.; et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: Recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 2018, 20, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Trivedi, D.V.; Sarkar, S.S.; Adhikari, A.S.; Sunitha, M.S.; Sutton, S.; Ruppel, K.M.; Spudich, J.A. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat. Struct. Mol. Biol. 2017, 24, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Spudich, J.A. The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 2015, 43, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegrave, M.; Peckham, M. Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat. Rec. 2014, 297, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Renaux, A.; Papadimitriou, S.; Versbraegen, N.; Nachtegael, C.; Boutry, S.; Nowe, A.; Smits, G.; Lenaerts, T. ORVAL: A novel platform for the prediction and exploration of disease-causing oligogenic variant combinations. Nucleic Acids Res. 2019, 47, W93–W98. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Variant | Associated Phenotype | Scaled CADD Score | SIFT Prediction | PolyPhen2 Prediction | GnomAD Genomes Allele Arequency | ALFA Allele Frequency |
|---|---|---|---|---|---|---|
| D3N | HLHS [63] | 25.6 | damaging | probably damaging | 9.20 × 10−5 | 4.00 × 10−5 |
| R17H | ASD [30] | 26.3 | damaging | probably damaging | 6.57 × 10−6 | not reported |
| P40R | CHD [23] | 20.6 | damaging | probably damaging | not reported | not reported |
| E98K | Shone complex [27], HLHS [63], CHD [26] | 27.4 | damaging | probably damaging | 3.15 × 10−4 | 6.12 × 10−4 |
| R108C | HLHS [63] | 29 | damaging | probably damaging | not reported | not reported |
| Y115N | HLHS [8] | 28.6 | damaging | probably damaging | not reported | not reported |
| S118L | HLHS [63] | 27.4 | damaging | possibly damaging | 6.60 × 10−6 | not reported |
| S180Y | DCM [52] | 26.4 | damaging | probably damaging | not reported | not reported |
| R204H | HCM [43] | 23.7 | tolerated | probably damaging | 4.60 × 10−5 | 2.20 × 10−5 |
| D208N | AVSD [24] | 22.3 | tolerated | benign | 4.15 × 10−3 | 6.38 × 10−3 |
| A230P | TA, LVH [26] | 25.4 | damaging | probably damaging | not reported | not reported |
| H252Q | TGA, PFO [26] | 22.4 | damaging | probably damaging | 2.63 × 10−5 | 6.00 × 10−5 |
| I275N | DCM [48,49] | 17.4 | damaging | probably damaging | 1.84 × 10−4 | 3.63 × 10−4 |
| Q277H | HLHS [8,63], CHD [23] | 20.9 | damaging | benign | 2.76 × 10−4 | 4.04 × 10−4 |
| E329stop | ASD [29] | 43 | damaging | N/A | not reported | not reported |
| D383N | HLHS [8] | 25.5 | damaging | probably damaging | not reported | not reported |
| S385L | HLHS [8] | 23.1 | tolerated | benign | 9.87 × 10−5 | not reported |
| L388F | Shone complex [27] | 24.7 | damaging | probably damaging | not reported | not reported |
| G415R | MVP [57] | 26.9 | damaging | probably damaging | 6.57 × 10−5 | 7.00 × 10−5 |
| M436V | HLHS [8] | 24.6 | tolerated | probably damaging | not reported | not reported |
| R443P | HLHS [8] | 27.2 | damaging | probably damaging | not reported | 3.00 × 10−5 |
| E501stop | TA [26] | 39 | damaging | N/A | not reported | not reported |
| I512T | Shone complex [26] | 23.2 | damaging | benign | 1.97 × 10−5 | 3.00 × 10−5 |
| E526K | ASD [64] | 27.5 | damaging | possibly damaging | 6.57 × 10−6 | not reported |
| C539R | ASD [30] | 26.3 | damaging | possibly damaging | not reported | not reported |
| K543R | ASD [30] | 25.4 | damaging | possibly damaging | 5.93 × 10−5 | 1.20 × 10−4 |
| R568C | PPCM [53], DCM [48] | 25.7 | damaging | probably damaging | 3.29 × 10−5 | 9.00 × 10−5 |
| G585S | Shone complex [27] | 24.4 | damaging | possibly damaging | 1.31 × 10−4 | 1.91 × 10−4 |
| D588A | HLHS [8,9,63] | 22.6 | tolerated | benign | 1.40 × 10−3 | 2.51 × 10−3 |
| V606I | HLHS [63] | 23.1 | damaging | benign | 6.57 × 10−5 | not reported |
| D629N | HCM [44] | 22.5 | tolerated | possibly damaging | 5.91 × 10−5 | 7.00 × 10−5 |
| F646L | HLHS [63] | 24.4 | damaging | possibly damaging | not reported | not reported |
| R654W | Arrhythmia [33] | 31 | damaging | probably damaging | 1.97 × 10−5 | 4.00 × 10−5 |
| N678S | ASD, TA [25] | 25.1 | damaging | probably damaging | not reported | 7.00 × 10−5 |
| V700M | PFO [26,65] | 26.8 | damaging | probably damaging | 6.57 × 10−6 | not reported |
| I704N | HLHS [9] | 26.7 | damaging | probably damaging | not reported | not reported |
| R721W | SSS [35], CoA [58] | 26 | damaging | probably damaging | 1.91 × 10−5 | not reported |
| R795Q | HCM [42] | 25.9 | damaging | probably damaging | not reported | 1.00 × 10−4 |
| R795W | HLHS [8] | 26 | damaging | probably damaging | 1.38 × 10−4 | 2.54 × 10−4 |
| I806T | DCM [41] | 22.6 | tolerated | benign | not reported | not reported |
| R809C | HCM [43] | 26 | damaging | probably damaging | 1.31 × 10−5 | 8.00 × 10−5 |
| I820N | ASD [31] | 26.3 | damaging | not reported | not reported | |
| P830L | DCM [47,50] | 26.3 | damaging | probably damaging | not reported | not reported |
| K849del | HLHS [8] | N/A | N/A | N/A | not reported | not reported |
| T864M | HLHS [63] | 20.7 | tolerated | benign | 7.89 × 10−5 | 1.40 × 10−4 |
| E885K | WPW [32] | 29.3 | damaging | probably damaging | not reported | not reported |
| A895V | CHD [26] | 32 | damaging | possibly damaging | 6.57 × 10−6 | not reported |
| E933del | SSS [39] | N/A | N/A | N/A | not reported | not reported |
| A936V | HLHS [8], AVSD [24] | 25.9 | tolerated | possibly damaging | 6.24 × 10−4 | 2.16 × 10−4 |
| E948K | MVP [57], HLHS [63] | 27.4 | damaging | probably damaging | 5.91 × 10−5 | 3.00 × 10−5 |
| C949stop | ARVC [54] | 36 | damaging | N/A | not reported | not reported |
| E951stop | ASD [29] | 38 | damaging | N/A | not reported | not reported |
| A964S | HLHS [8] | 26.3 | tolerated | 1.37 × 10−3 | 1.76 × 10−4 | |
| A1004S | ASD [30], DCM [47,48,50] | 23.6 | tolerated | benign | 1.10 × 10−3 | 1.03 × 10−3 |
| R1047C | DCM [51] | 29.7 | damaging | probably damaging | 7.23 × 10−5 | 2.00 × 10−5 |
| R1052stop | MVP [57] | 38 | damaging | N/A | 1.97 × 10−5 | 3.00 × 10−5 |
| Q1065H | HCM [47] | 23.5 | damaging | probably damaging | 1.51 × 10−4 | 1.10 × 10−4 |
| I1068T | Shone complex [27] | 21.9 | tolerated | benign | not reported | not reported |
| R1116S | ASD [26] | 24.6 | damaging | probably damaging | 7.90 × 10−5 | 7.00 × 10−5 |
| R1116C | HCM [66] | 26 | damaging | probably damaging | not reported | not reported |
| R1116H | SSS [67] | 25 | damaging | probably damaging | 4.60 × 10−5 | 7.00 × 10−5 |
| R1151Q | HLHS [8] | 28.9 | tolerated | probably damaging | 1.32 × 10−5 | not reported |
| R1177W | DCM [48] | 25.4 | damaging | probably damaging | 2.64 × 10−5 | 4.00 × 10−5 |
| T1190I | HLHS [63] | 25.4 | damaging | probably damaging | 3.95 × 10−5 | not reported |
| E1207K | HLHS [9] | 27.3 | damaging | probably damaging | 5.93 × 10−5 | 5.00 × 10−5 |
| R1252Q | SSS [67] | 24.9 | damaging | not reported | not reported | |
| T1253M | DCM [51] | 25.3 | tolerated | probably damaging | 1.31 × 10−5 | not reported |
| R1279stop | ASD [28] | 38 | damaging | N/A | not reported | not reported |
| R1291P | HLHS [63] | 31 | damaging | probably damaging | not reported | not reported |
| A1298V | HLHS [8] | 25 | tolerated | possibly damaging | 1.12 × 10−4 | 2.00 × 10−4 |
| K1307M | AF [34] | 28.8 | damaging | probably damaging | not reported | not reported |
| D1316E | SCD [37] | 19.7 | tolerated | possibly damaging | 1.31 × 10−5 | 3.00 × 10−5 |
| E1323V | AF [34] | 31 | damaging | probably damaging | not reported | not reported |
| A1327V | Shone complex [27] | 28.1 | damaging | probably damaging | 6.21 × 10−4 | 1.70 × 10−4 |
| S1337L | LVNC [55] | 28.9 | damaging | probably damaging | 1.98 × 10−5 | 4.00 × 10−5 |
| A1366D | SDK, SAR, PFO, AS [26] | 28.9 | damaging | probably damaging | not reported | not reported |
| T1379M | HLHS [8,9], MVP [57], CHD [26] | 27.3 | damaging | probably damaging | 3.22 × 10−4 | 5.87 × 10−4 |
| R1398Q | HLHS [63], CHD [23] | 23.6 | damaging | benign | 4.14 × 10−4 | 5.26 × 10−4 |
| A1440P | DCM [48] | 26.4 | tolerated | possibly damaging | not reported | not reported |
| A1443D | ASD [26,65], HLHS [8,63] | 26.3 | damaging | 1.71 × 10−4 | 1.30 × 10−4 | |
| E1457K | DCM [47,50] | 28.1 | damaging | probably damaging | 1.31 × 10−5 | not reported |
| R1502Q | DCM [48,49] | 30 | damaging | probably damaging | 2.17 × 10−4 | 2.61 × 10−4 |
| E1503V | HLHS [8] | 33 | damaging | probably damaging | not reported | not reported |
| E1584K | HLHS [8] | 28.7 | damaging | probably damaging | 2.63 × 10−5 | not reported |
| R1608C | CHD [23] | 26 | damaging | probably damaging | 4.60 × 10−5 | 8.00 × 10−5 |
| R1610C | Shone complex [27] | 28.7 | damaging | probably damaging | 7.89 × 10−5 | 6.00 × 10−5 |
| A1674T | CHD [41] | 24.5 | tolerated | benign | 4.60 × 10−5 | 3.00× 10−5 |
| E1713K | HLHS [63] | 30 | damaging | probably damaging | 5.26 × 10−5 | not reported |
| E1754stop | HLHS [8] | 45 | damaging | N/A | 1.97 × 10−5 | not reported |
| E1827D | HLHS [63] | 25.1 | damaging | probably damaging | not reported | not reported |
| K1840R | HLHS [8] HCM [43] | 25.1 | tolerated | probably damaging | 1.12 × 10−4 | 2.50 × 10−4 |
| D1859N | HLHS [63] | 29.8 | damaging | probably damaging | 1.94 × 10−5 | not reported |
| R1865Q | DIVC, ASD, VSD [26] | 29.8 | damaging | probably damaging | 4.06 × 10−5 | 3.00 × 10−5 |
| A1891T | Shone complex [27] | 26.8 | damaging | probably damaging | not reported | not reported |
| R1899C | DCM [48] | 32 | damaging | probably damaging | 1.31 × 10−5 | not reported |
| R1899H | Shone complex [27] | 31 | damaging | probably damaging | 3.94 × 10−5 | 3.00 × 10−5 |
| R1911P | HLHS [63] | 29.6 | damaging | probably damaging | not reported | not reported |
| K1932stop | Shone complex [27] | 51 | damaging | N/A | 1.97 × 10−5 | not reported |
| Model System | MYH6 Variant | Disease Association | Cellular Phenotype | Reference |
|---|---|---|---|---|
| Purified recombinant protein | I820N | ASD |
| [31] |
| HeLa cells | E933del | SSS |
| [39] |
| Myofibrils differentiated from C2C12 myoblasts | A230P | TA, ASD, LVH |
| [26] |
| A1366D | AS, SDK, SAR, PFO |
| [26] | |
| H252Q | TGA, PFO |
| [26] | |
| V700M | PFO |
| [26] | |
| E526K | ASD |
| [64] | |
| R1822_ E1823dup | ASD |
| [73] | |
| Rat or mouse ventricular myocytes expressing human MYH6 | R721W | SSS, CoA |
| [39] |
| P830L | DCM |
| [74] | |
| E933del | SSS |
| [39] | |
| A1004S | ASD, DCM |
| [74] | |
| Induced pluripotent stem-cell-derived cardiomyocytes (iPSC-CMs) | R443P | HLHS |
| [8,75] |
| Patient cardiac tissue | R443P | HLHS |
| [8] |
| N598fs | ACM |
| [54] | |
| D629N | HCM |
| [44] | |
| A822T | SCD |
| [36] | |
| K849del | HLHS |
| [8,75] | |
| E1503V | HLHS |
| [75] | |
| S385L & M436V | HLHS |
| [75] | |
| Zebrafish | E933del | SSS |
| [39] |
| R1252Q | SSS |
| [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anfinson, M.; Fitts, R.H.; Lough, J.W.; James, J.M.; Simpson, P.M.; Handler, S.S.; Mitchell, M.E.; Tomita-Mitchell, A. Significance of α-Myosin Heavy Chain (MYH6) Variants in Hypoplastic Left Heart Syndrome and Related Cardiovascular Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 144. https://doi.org/10.3390/jcdd9050144
Anfinson M, Fitts RH, Lough JW, James JM, Simpson PM, Handler SS, Mitchell ME, Tomita-Mitchell A. Significance of α-Myosin Heavy Chain (MYH6) Variants in Hypoplastic Left Heart Syndrome and Related Cardiovascular Diseases. Journal of Cardiovascular Development and Disease. 2022; 9(5):144. https://doi.org/10.3390/jcdd9050144
Chicago/Turabian StyleAnfinson, Melissa, Robert H. Fitts, John W. Lough, Jeanne M. James, Pippa M. Simpson, Stephanie S. Handler, Michael E. Mitchell, and Aoy Tomita-Mitchell. 2022. "Significance of α-Myosin Heavy Chain (MYH6) Variants in Hypoplastic Left Heart Syndrome and Related Cardiovascular Diseases" Journal of Cardiovascular Development and Disease 9, no. 5: 144. https://doi.org/10.3390/jcdd9050144
APA StyleAnfinson, M., Fitts, R. H., Lough, J. W., James, J. M., Simpson, P. M., Handler, S. S., Mitchell, M. E., & Tomita-Mitchell, A. (2022). Significance of α-Myosin Heavy Chain (MYH6) Variants in Hypoplastic Left Heart Syndrome and Related Cardiovascular Diseases. Journal of Cardiovascular Development and Disease, 9(5), 144. https://doi.org/10.3390/jcdd9050144