
Understanding the Role of SERCA2a Microdomain Remodeling in Heart Failure Induced by Obesity and Type 2 Diabetes


{kind=link}
Abstract
:1. Introduction
2. Obesity and T2D
3. Heart Failure Induced by Obesity and T2D
4. cAMP and Its Microdomains
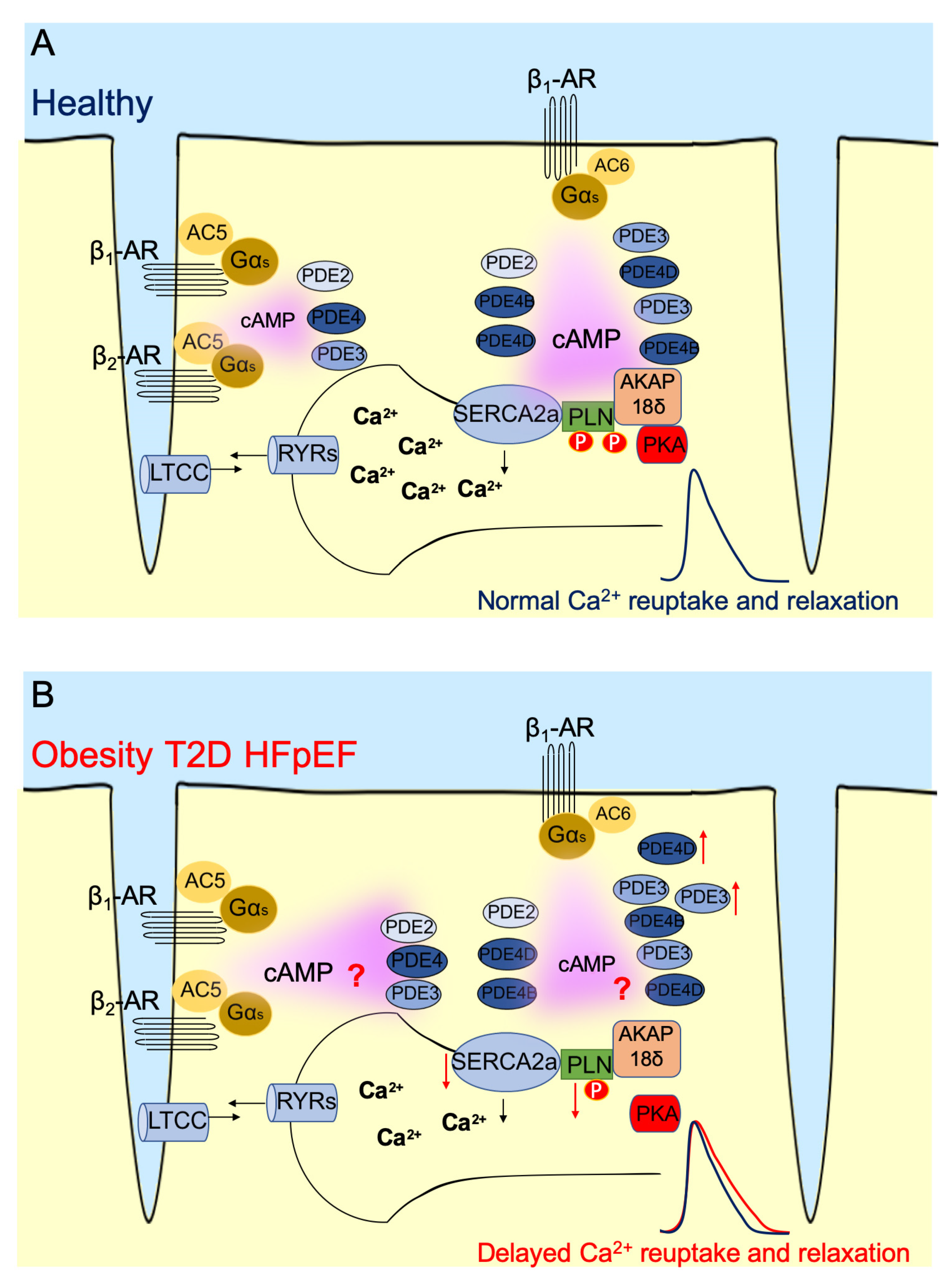
5. PLN/SERCA2a Microdomain in Heart
6. PLN/SERCA2a Microdomain in Heart Dysfunction Induced by Obesity and T2D
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef] [PubMed]
- Despres, J.P. Body fat distribution and risk of cardiovascular disease: An update. Circulation 2012, 126, 1301–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldeyer, R.; Brinks, R.; Rathmann, W.; Giani, G.; Icks, A. Projection of the burden of type 2 diabetes mellitus in Germany: A demographic modelling approach to estimate the direct medical excess costs from 2010 to 2040. Diabet. Med. 2013, 30, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, E.A.; Khavjou, O.A.; Thompson, H.; Trogdon, J.G.; Pan, L.; Sherry, B.; Dietz, W. Obesity and severe obesity forecasts through 2030. Am. J. Prev. Med. 2012, 42, 563–570. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Kajio, H. Beta-blocker use and cardiovascular event risk in patients with heart failure with preserved ejection fraction. Sci. Rep. 2018, 8, 9556. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Kajio, H. Abdominal Obesity Is Associated with an Increased Risk of All-Cause Mortality in Patients with HFpEF. J. Am. Coll. Cardiol. 2017, 70, 2739–2749. [Google Scholar] [CrossRef]
- Zaccolo, M. cAMP signal transduction in the heart: Understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 2009, 158, 50–60. [Google Scholar] [CrossRef] [Green Version]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef]
- Gonzalez-Muniesa, P.; Martinez-Gonzalez, M.A.; Hu, F.B.; Despres, J.P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Primers 2017, 3, 17034. [Google Scholar] [CrossRef]
- World Health Organization. Obesity: Preventing and Managing the Global Epidemic: Report of aWHO Consultation; WHO Technical Report Series; World Health Organization: Geneva, Switerzerland, 2000; p. 9. [Google Scholar]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- World Health Organization. Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. REPORT of a WHO Consultation, Part. 1: Diagnosis and Classification of Diabetes Mellitus; World Health Organization: Geneva, Switerzerland, 1999. [Google Scholar]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Piche, M.E.; Tchernof, A.; Despres, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Lyass, A.; Enserro, D.; Larson, M.G.; Ho, J.E.; Kizer, J.R.; Gottdiener, J.S.; Psaty, B.M.; Vasan, R.S. Temporal Trends in the Incidence of and Mortality Associated with Heart Failure with Preserved and Reduced Ejection Fraction. JACC Heart Fail. 2018, 6, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart Failure with Preserved Ejection Fraction In Perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.J.; Borlaug, B.A.; Kitzman, D.W.; McCulloch, A.D.; Blaxall, B.C.; Agarwal, R.; Chirinos, J.A.; Collins, S.; Deo, R.C.; Gladwin, M.T.; et al. Research Priorities for Heart Failure with Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 2020, 141, 1001–1026. [Google Scholar] [CrossRef]
- Boyle, J.P.; Thompson, T.J.; Gregg, E.W.; Barker, L.E.; Williamson, D.F. Projection of the year 2050 burden of diabetes in the US adult population: Dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul. Health Metr. 2010, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.K. Epidemiology of Heart Failure with Preserved Ejection Fraction in Pediatric Population with Obesity. Pharmacoepidemiol. Drug Saf. 2020, 29, 455. [Google Scholar]
- Wheeler, D.C.; Valensi, P.; Sousa-Uva, M.; Seferović, P.M.; Sattar, N.; Roffi, M.; Rocca, B.; Östgren, C.J.; Mellbin, L.G.; Marx, N.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar]
- Libby, P.; Theroux, P. Pathophysiology of coronary artery disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef] [Green Version]
- Irie, J.; Itoh, H. Dysbiosis in the Pathophysiology of Coronary Artery Disease. J. Atheroscler. Thromb. 2016, 23, 901–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Girgis, S.; Hassan, A.; Rudick, S.; Becker, R.C. Inflammation and coronary artery disease: From pathophysiology to Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Coron. Artery Dis. 2018, 29, 429–437. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Silverman, D.N.; Plante, T.B.; Infeld, M.; Callas, P.W.; Juraschek, S.P.; Dougherty, G.B.; Meyer, M. Association of beta-Blocker Use with Heart Failure Hospitalizations and Cardiovascular Disease Mortality among Patients with Heart Failure with a Preserved Ejection Fraction: A Secondary Analysis of the TOPCAT Trial. JAMA Netw. Open 2019, 2, e1916598. [Google Scholar] [CrossRef]
- Sakata, Y.; Shiba, N.; Takahashi, J.; Miyata, S.; Nochioka, K.; Miura, M.; Takada, T.; Saga, C.; Shinozaki, T.; Sugi, M.; et al. Clinical impacts of additive use of olmesartan in hypertensive patients with chronic heart failure: The supplemental benefit of an angiotensin receptor blocker in hypertensive patients with stable heart failure using olmesartan (SUPPORT) trial. Eur. Heart J. 2015, 36, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.J.; Lam, C.S.P.; Svedlund, S.; Saraste, A.; Hage, C.; Tan, R.S.; Beussink-Nelson, L.; Ljung Faxen, U.; Fermer, M.L.; Broberg, M.A.; et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur. Heart J. 2018, 39, 3439–3450. [Google Scholar] [CrossRef]
- Suhrs, H.E.; Schroder, J.; Bove, K.B.; Mygind, N.D.; Frestad, D.; Michelsen, M.M.; Lange, T.; Gustafsson, I.; Kastrup, J.; Prescott, E. Inflammation, non-endothelial dependent coronary microvascular function and diastolic function-Are they linked? PLoS ONE 2020, 15, e0236035. [Google Scholar] [CrossRef]
- Ko, K.Y.; Wu, Y.W.; Liu, C.W.; Cheng, M.F. Longitudinal evaluation of myocardial glucose metabolism and contractile function in obese type 2 diabetic dbdb mice using small-animal dynamic 18F-FDG PET and echocardiography. Oncotarget 2017, 8, 87795–87808. [Google Scholar] [CrossRef] [Green Version]
- Ceylan-Isik, A.F.; Sreejayan, N.; Ren, J. Endoplasmic reticulum chaperon tauroursodeoxycholic acid alleviates obesity-induced myocardial contractile dysfunction. J. Mol. Cell Cardiol. 2011, 50, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Szokol, M.; Priksz, D.; Bombicz, M.; Varga, B.; Kovacs, A.; Fulop, G.A.; Csipo, T.; Posa, A.; Toth, A.; Papp, Z.; et al. Long Term Osmotic Mini Pump Treatment with Alpha-MSH Improves Myocardial Function in Zucker Diabetic Fatty Rats. Molecules 2017, 22, 1702. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.; Norton, G.R.; Woodiwiss, A.J.; Lochner, A.; du Toit, E.F. Dependence of Cardiac Systolic Function on Elevated Fatty Acid Availability in Obese, Insulin-Resistant Rats. J. Card. Fail. 2016, 22, 560–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Rajesh, M.; Zhang, J.; Zhou, S.; Wang, S.; Sun, J.; Tan, Y.; Zheng, Y.; Cai, L. Protection by dimethyl fumarate against diabetic cardiomyopathy in type 1 diabetic mice likely via activation of nuclear factor erythroid-2 related factor 2. Toxicol Lett. 2018, 287, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Nikolaev, V.O. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol. 2013, 207, 650–662. [Google Scholar] [CrossRef] [PubMed]
- De Jong, K.A.; Nikolaev, V.O. Multifaceted remodelling of cAMP microdomains driven by different aetiologies of heart failure. FEBS J. 2021, 288, 6603–6622. [Google Scholar] [CrossRef] [PubMed]
- Fischmeister, R.; Castro, L.R.; Abi-Gerges, A.; Rochais, F.; Jurevicius, J.; Leroy, J.; Vandecasteele, G. Compartmentation of cyclic nucleotide signaling in the heart: The role of cyclic nucleotide phosphodiesterases. Circ. Res. 2006, 99, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Sato, P.Y.; Chuprun, J.K.; Schwartz, M.; Koch, W.J. The evolving impact of g protein-coupled receptor kinases in cardiac health and disease. Physiol. Rev. 2015, 95, 377–404. [Google Scholar] [CrossRef] [Green Version]
- Froese, A.; Nikolaev, V.O. Imaging alterations of cardiomyocyte cAMP microdomains in disease. Front. Pharmacol. 2015, 6, 172. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, K.; Zaccolo, M. Using cAMP Sensors to Study Cardiac Nanodomains. J. Cardiovasc. Dev. Dis. 2018, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef] [Green Version]
- Primeau, J.O.; Armanious, G.P.; Fisher, M.E.; Young, H.S. The SarcoEndoplasmic Reticulum Calcium ATPase. Subcell Biochem. 2018, 87, 229–258. [Google Scholar]
- Gorski, P.A.; Ceholski, D.K.; Young, H.S. Structure-Function Relationship of the SERCA Pump and Its Regulation by Phospholamban and Sarcolipin. Adv. Exp. Med. Biol. 2017, 981, 77–119. [Google Scholar] [PubMed]
- Tsai, C.T.; Wu, C.K.; Lee, J.K.; Chang, S.N.; Kuo, Y.M.; Wang, Y.C.; Lai, L.P.; Chiang, F.T.; Hwang, J.J.; Lin, J.L. TNF-alpha down-regulates sarcoplasmic reticulum Ca(2)(+) ATPase expression and leads to left ventricular diastolic dysfunction through binding of NF-kappaB to promoter response element. Cardiovasc. Res. 2015, 105, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, D.S.; Carnes, C.A.; Abraham, W.T.; Bristow, M.R. Mechanisms of disease: Beta-adrenergic receptors--alterations in signal transduction and pharmacogenomics in heart failure. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, R.R.; Lingam, S.J.; Wang, H.Y.; Bollen, I.A.; Hughes, G.; Galvin, I.F.; Bunton, R.W.; Bahn, A.; Katare, R.; Baldi, J.C.; et al. Impaired relaxation despite upregulated calcium-handling protein atrial myocardium from type 2 diabetic patients with preserved ejection fraction. Cardiovasc. Diabetol. 2014, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Kirchberger, M.A.; Katz, A.M. Phosphorylation of a 22,000-dalton component of the cardiac sarcoplasmic reticulum by adenosine 3′:5′-monophosphate-dependent protein kinase. J. Biol. Chem. 1975, 250, 2640–2647. [Google Scholar] [CrossRef]
- Tsuji, T.; Del Monte, F.; Yoshikawa, Y.; Abe, T.; Shimizu, J.; Nakajima-Takenaka, C.; Taniguchi, S.; Hajjar, R.J.; Takaki, M. Rescue of Ca2+ overload-induced left ventricular dysfunction by targeted ablation of phospholamban. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H310–H317. [Google Scholar] [CrossRef]
- Glaves, J.P.; Primeau, J.O.; Espinoza-Fonseca, L.M.; Lemieux, M.J.; Young, H.S. The Phospholamban Pentamer Alters Function of the Sarcoplasmic Reticulum Calcium Pump SERCA. Biophys. J. 2019, 116, 633–647. [Google Scholar] [CrossRef] [Green Version]
- James, P.; Inui, M.; Tada, M.; Chiesi, M.; Carafoli, E. Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature 1989, 342, 90–92. [Google Scholar] [CrossRef]
- Haghighi, K.; Kolokathis, F.; Pater, L.; Lynch, R.A.; Asahi, M.; Gramolini, A.O.; Fan, G.-C.; Tsiapras, D.; Hahn, H.S.; Adamopoulos, S.; et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Investig. 2003, 111, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, J.P.; Kamisago, M.; Asahi, M.; Li, G.H.; Ahmad, F.; Mende, U.; Kranias, E.G.; MacLennan, D.H.; Seidman, J.G.; Seidman, C.E. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003, 299, 1410–1413. [Google Scholar] [CrossRef]
- Nelson, S.E.D.; Ha, K.N.; Gopinath, T.; Exline, M.H.; Mascioni, A.; Thomas, D.D.; Veglia, G. Effects of the Arg9Cys and Arg25Cys mutations on phospholamban’s conformational equilibrium in membrane bilayers. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Kadambi, V.; Schmidt, A.G.; Tepe, N.M.; Biniakiewicz, D.; Gerst, M.J.; Canning, A.M.; Abraham, W.T.; Hoit, B.D.; Liggett, S.B.; et al. Interactions between phospholamban and beta-adrenergic drive may lead to cardiomyopathy and early mortality. Circulation 2001, 103, 889–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belke, D.D.; Swanson, E.A.; Dillmann, W.H. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 2004, 53, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Wold, L.E.; Dutta, K.; Mason, M.M.; Ren, J.; Cala, S.E.; Schwanke, M.L.; Davidoff, A.J. Impaired SERCA function contributes to cardiomyocyte dysfunction in insulin resistant rats. J. Mol. Cell Cardiol. 2005, 39, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Vasanji, Z.; Cantor, E.J.; Juric, D.; Moyen, M.; Netticadan, T. Alterations in cardiac contractile performance and sarcoplasmic reticulum function in sucrose-fed rats is associated with insulin resistance. Am. J. Physiol. Cell Physiol. 2006, 291, C772–C780. [Google Scholar] [CrossRef] [Green Version]
- Ouwens, D.M.; Boer, C.; Fodor, M.; de Galan, P.; Heine, R.J.; Maassen, J.A.; Diamant, M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia 2005, 48, 1229–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.Y.; Liu, F.C.; Deng, C.Y.; Zhang, M.Z.; Yang, M.; Xiao, D.Z.; Lin, Q.X.; Cai, S.T.; Kuang, S.J.; Chen, J.; et al. Left ventricular deformation associated with cardiomyocyte Ca(2+) transients delay in early stage of low-dose of STZ and high-fat diet induced type 2 diabetic rats. BMC Cardiovasc. Disord 2016, 16, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balderas-Villalobos, J.; Molina-Munoz, T.; Mailloux-Salinas, P.; Bravo, G.; Carvajal, K.; Gomez-Viquez, N.L. Oxidative stress in cardiomyocytes contributes to decreased SERCA2a activity in rats with metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1344–H1353. [Google Scholar] [CrossRef] [Green Version]
- Abdurrachim, D.; Ciapaite, J.; Wessels, B.; Nabben, M.; Luiken, J.J.; Nicolay, K.; Prompers, J.J. Cardiac diastolic dysfunction in high-fat diet fed mice is associated with lipotoxicity without impairment of cardiac energetics in vivo. Biochim. Biophys. Acta 2014, 1842, 1525–1537. [Google Scholar] [CrossRef]
- Lima-Leopoldo, A.P.; Leopoldo, A.S.; da Silva, D.C.; do Nascimento, A.F.; de Campos, D.H.; Luvizotto, R.A.; de Deus, A.F.; Freire, P.P.; Medeiros, A.; Okoshi, K.; et al. Long-term obesity promotes alterations in diastolic function induced by reduction of phospholamban phosphorylation at serine-16 without affecting calcium handling. J. Appl. Physiol. 2014, 117, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.C.; Chang, W.T.; Kuo, F.Y.; Chen, Z.C.; Li, Y.; Cheng, J.T. TGR5 activation ameliorates hyperglycemia-induced cardiac hypertrophy in H9c2 cells. Sci. Rep. 2019, 9, 3633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, Y.; Fu, Q.; Xu, B.; Zhang, Y.; Kim, S.; Tan, R.; Barbagallo, F.; West, T.; Anderson, E.; et al. Inhibiting Insulin-Mediated beta2-Adrenergic Receptor Activation Prevents Diabetes-Associated Cardiac Dysfunction. Circulation 2017, 135, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Okatan, E.N.; Turan, B. The contribution of phosphodiesterases to cardiac dysfunction in rats with metabolic syndrome induced by a high-carbohydrate diet. Can. J. Physiol. Pharmacol. 2019, 97, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, P.; Nikolaev, V.O.; De Jong, K.A. Understanding the Role of SERCA2a Microdomain Remodeling in Heart Failure Induced by Obesity and Type 2 Diabetes. J. Cardiovasc. Dev. Dis. 2022, 9, 163. https://doi.org/10.3390/jcdd9050163
Lai P, Nikolaev VO, De Jong KA. Understanding the Role of SERCA2a Microdomain Remodeling in Heart Failure Induced by Obesity and Type 2 Diabetes. Journal of Cardiovascular Development and Disease. 2022; 9(5):163. https://doi.org/10.3390/jcdd9050163
Chicago/Turabian StyleLai, Ping, Viacheslav O. Nikolaev, and Kirstie A. De Jong. 2022. "Understanding the Role of SERCA2a Microdomain Remodeling in Heart Failure Induced by Obesity and Type 2 Diabetes" Journal of Cardiovascular Development and Disease 9, no. 5: 163. https://doi.org/10.3390/jcdd9050163