
The Role of Epicardial Adipose Tissue in the Development of Atrial Fibrillation, Coronary Artery Disease and Chronic Heart Failure in the Context of Obesity and Type 2 Diabetes Mellitus: A Narrative Review


Abstract
:1. Introduction
2. Physiology of Epicardial Adipose Tissue
2.1. Origin, Distribution, and Anatomy of EAT
2.2. Brown vs. White Adipose Tissue and Cardiometabolic Modulation
2.3. EAT Secretome and Adipokines
2.4. Diagnostic Imaging Characterizing the Thickness and Volume of EAT
3. Pathophysiologic States of Epicardial Adipose Tissue
3.1. Epicardial Adipose Tissue and Atrial Fibrillation
3.2. Epicardial Adipose Tissue and Coronary Artery Disease
3.3. Epicardial Adipose Tissue and Chronic Heart Failure
4. Clinical Implications and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AF = atrial fibrillation |
| AGE = advanced glycation end products |
| AP = action potential |
| CAC = coronary artery calcium |
| CAD = coronary artery disease |
| CD = cluster of differentiation |
| CHF = chronic heart failure |
| CT = computed tomography |
| CVD = cardiovascular disease |
| Cx = connexins |
| EAD/DAD = early/delayed after depolarizations |
| EAT = epicardial adipose tissue |
| ECM = extracellular matrix |
| EF = ejection fraction |
| EFV = epicardial fat volume |
| FFA = free fatty acids |
| HDL = high-density lipoprotein |
| IL = interleukin |
| LAA/RAA = left/right atrial appendages |
| LDL = low-density lipoprotein |
| LV/LA = left ventricle/atrium |
| MMP = matrix metalloproteinases |
| MS = metabolic syndrome |
| NEFA = non-esterified fatty acids |
| NLRP3 = leucine-rich repeat-containing receptor family pyrin domain-containing 3 |
| PVAT = perivascular adipose tissue |
| SD = standard deviation |
| T2DM = type 2 diabetes mellitus |
| TGF = transforming growth factor |
| TNF = tumor necrosis factor |
References
- Purnell, J.Q. Definitions, Classification, and Epidemiology of Obesity. [Updated 12 April 2018]. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000; Available online: https://www.ncbi.nlm.nih.gov/books/NBK279167/ (accessed on 25 June 2021).
- WHO. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 28 May 2021).
- Apovian, C.M.; Okemah, J.; O’Neil, P.M. Body Weight Considerations in the Management of Type 2 Diabetes. Adv. Ther. 2019, 36, 44–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Cardiovascular Diseases. 2021. Available online: https://www.who.int/health-topics/cardiovascular-diseases/#tab=tab_1 (accessed on 25 May 2021).
- Algoblan, A.; Alalfi, M.; Khan, M. Mechanism Linking Diabetes Mellitus and Obesity. Diabetes Metab. Syndr. Obes. 2014, 7, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pippitt, K.; Li, M.; Gurgle, H.E. Diabetes Mellitus: Screening and Diagnosis. Am. Fam. Physician 2016, 93, 103–109; Erratum in Am. Fam. Physician 2016, 94, 533. [Google Scholar]
- Heiss, G.; Snyder, M.; Teng, Y.; Schneiderman, N.; Llabre, M.M.; Cowie, C.; Carnethon, M.; Kaplan, R.; Giachello, A.; Gallo, L.; et al. Prevalence of Metabolic Syndrome Among Hispanics/Latinos of Diverse Background: The Hispanic Community Health Study/Study of Latinos. Diabetes Care 2014, 37, 2391–2399. [Google Scholar] [CrossRef] [Green Version]
- Roden, M.; Price, T.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of Free Fatty Acid-induced Insulin Resistance in Humans. J. Clin. Investig. 1996, 97, 2859–2865. [Google Scholar] [CrossRef] [Green Version]
- Matheus, A.; Tannus, L.; Cobas, R.; Palma, C.; Negrato, C.; Gomes, M. Impact of Diabetes on Cardiovascular Disease: An Update. Int. J. Hypertens. 2013, 2013, 653789. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G. Anatomy of the Epicardial Adipose Tissue. In Contemporary Cardiology; Humana: Cham, Switzerland, 2020; pp. 1–8. [Google Scholar] [CrossRef]
- Antonopoulos, A.; Antoniades, C. The Role of Epicardial Adipose Tissue in Cardiac Biology: Classic Concepts and Emerging Roles. J. Physiol. 2017, 595, 3907–3917. [Google Scholar] [CrossRef]
- Selthofer-Relatić, K.; Bošnjak, I. Myocardial Fat as a Part of Cardiac Visceral Adipose Tissue: Physiological and Pathophysiological View. J. Endocrinol. Investig. 2015, 38, 933–939. [Google Scholar] [CrossRef]
- Newman, T. Pericarditis: Symptoms, Diagnosis, and Treatment. 2018. Available online: https://www.medicalnewstoday.com/articles/193320 (accessed on 16 May 2022).
- Betts, J.; Young, K.; James, W. Anatomy and Physiology. 2013. Available online: https://openstax.org/books/anatomy-and-physiology/pages/1-introduction (accessed on 16 May 2022).
- Sacks, H.S.; Fain, J.N. Human Epicardial Fat: What is New and What is Missing? Clin. Exp. Pharmacol. Physiol. 2011, 38, 879–887. [Google Scholar] [CrossRef]
- Cherian, S.; Lopaschuk, G.; Carvalho, E. Cellular Cross-talk Between Epicardial Adipose Tissue and Myocardium in Relation to the Pathogenesis of Cardiovascular Disease. Am. J. Physiol. Endocrinol. Metabol. 2012, 303, E937–E949. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.; Chilton, E.; Raman, J.; Saxena, P.; McFarlane, C.; Trollope, A.F.; Kinobe, R.; Chilton, L. Are Interactions Between Epicardial Adipose Tissue, Cardiac Fibroblasts and Cardiac Myocytes Instrumental in Atrial Fibrosis and Atrial Fibrillation? Cells 2021, 10, 2501. [Google Scholar] [CrossRef]
- Aldiss, P.; Davies, G.; Woods, R.; Budge, H.; Sacks, H.; Symonds, M. ‘Browning’ the Cardiac and Peri-Vascular Adipose Tissues to Modulate Cardiovascular Risk. Int. J. Cardiol. 2017, 228, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G. Aging Effects on Epicardial Adipose Tissue. Front. Aging 2021, 2, 666260. [Google Scholar] [CrossRef]
- Iacobellis, G.; Bianco, A. Epicardial Adipose Tissue: Emerging Physiological, Pathophysiological and Clinical Features. Trends Endocrinol. Metabol. 2011, 22, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Tomášová, P.; Čermáková, M.; Pelantová, H.; Vecka, M.; Kratochvílová, H.; Lipš, M.; Lindner, J.; Ivák, P.; Netuka, I.; Šedivá, B.; et al. Lipid Profiling in Epicardial and Subcutaneous Adipose Tissue of Patients with Coronary Artery Disease. J. Proteome Res. 2020, 19, 3993–4003. [Google Scholar] [CrossRef]
- Christensen, R.; von Scholten, B.; Lehrskov, L.L.; Rossing, P.; Jørgensen, P.G. Epicardial Adipose Tissue: An Emerging Biomarker of Cardiovascular Complications in Type 2 Diabetes? Ther. Adv. Endocrinol. Metabol. 2020, 11, 2042018820928824. [Google Scholar] [CrossRef]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.; Diehl, J.; Arafat, H. Human Epicardial Adipose Tissue Is a Source of Inflammatory Mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, E.; Antoniades, C. The Role of Adipose Tissue in Cardiovascular Health and Disease. Nat. Rev. Cardiol. 2018, 16, 83–99. [Google Scholar] [CrossRef]
- Salazar, J.; Luzardo, E.; Mejías, J.; Rojas, J.; Ferreira, A.; Rivas-Ríos, J.; Bermúdez, V. Epicardial Fat: Physiological, Pathological, and Therapeutic Implications. Cardiol. Res. Pract. 2016, 2016, 1291537. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.; Shah, S.; Verma, S.; Oudit, G. Epicardial Adipose Tissue as a Metabolic Transducer: Role in Heart Failure and Coronary Artery Disease. Heart Fail. Rev. 2017, 22, 889–902. [Google Scholar] [CrossRef]
- Chao, T.-F.; Hung, C.-L.; Tsao, H.-M.; Lin, Y.-J.; Yun, C.-H.; Lai, Y.-H.; Chang, S.-L.; Lo, L.-W.; Hi, Y.-F.; Tuan, T.-A.; et al. Epicardial Adipose Tissue Thickness and Ablation Outcome of Atrial Fibrillation. PLoS ONE 2013, 8, e74926. [Google Scholar] [CrossRef]
- Kang, J.; Kim, Y.-C.; Park, J.J.; Kim, S.; Kang, S.-H.; Cho, Y.J.; Yoon, Y.E.; Oh, I.-Y.; Yoon, C.-H.; Suh, J.-W.; et al. Increased Epicardial Adipose Tissue Thickness is a Predictor of New-onset Diabetes Mellitus in Patients with Coronary Artery Disease Treated with High-Intensity Statins. Cardiovasc. Diabetol. 2018, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Milanese, G.; Silva, M.; Bruno, L.; Rossi, E.; Ferrari, C.; Grutta, L.; Maffei, E.; Toia, P.; Forte, E.; Bonadonna, R.C.; et al. Quantification of Epicardial Fat with Cardiac CT Angiography and Association with Cardiovascular Risk Factors in Symptomatic Patients: From the ALTER-BIO (Alternative Cardiovascular Bio-Imaging markers) Registry. Diagn. Interv. Radiol. 2019, 25, 35–41. [Google Scholar] [CrossRef]
- Rabkin, S. The Relationship Between Epicardial Fat and Indices of Obesity and the Metabolic Syndrome: A Systematic Review and Meta-Analysis. Metab. Syndr. Relat. Disord. 2014, 12, 31–42. [Google Scholar] [CrossRef]
- Shin, J.; Lee, J.; Lim, S.; Ha, H.S.; Kwon, H.S.; Park, Y.M.; Lee, W.C.; Kang, M.I.; Yim, H.W.; Yoon, K.H.; et al. Metabolic Syndrome as a Predictor of Type 2 Diabetes, and its Clinical Interpretations and Usefulness. J. Diabetes Investig. 2013, 4, 334–343. [Google Scholar] [CrossRef]
- Verma, B.; Katyal, D.; Patel, A.; Singh, V.R.; Kumar, S. Relation of systolic and diastolic epicardial adipose tissue thickness with presence and severity of coronary artery disease (The EAT CAD study). J. Fam. Med. Prim. Care 2019, 8, 1470–1475. [Google Scholar] [CrossRef]
- Mazurek, T.; Kobylecka, M.; Zielenkiewicz, M.; Kurek, A.; Kochman, J.; Filipiak, K.; Mazurek, K.; Huczek, Z.; Królicki, L.; Opolski, G. PET/CT Evaluation of 18F-FDG Uptake in Pericoronary Adipose Tissue in Patients with Stable Coronary Artery Disease: Independent Predictor of Atherosclerotic Lesions’ Formation? J. Nucl. Cardiol. 2016, 24, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Ernault, A.; Meijborg, V.; Coronel, R. Modulation of Cardiac Arrhythmogenesis by Epicardial Adipose Tissue. J. Am. Coll. Cardiol. 2021, 78, 1730–1745. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, A.; Khavandi, K.; Withers, S.; Sonoyama, K.; Clancy, O.; Jeziorska, M.; Laing, I.; Yates, A.P.; Pemberton, P.W.; Malik, R.A.; et al. Local Inflammation and Hypoxia Abolish the Protective Anticontractile Properties of Perivascular Fat in Obese Patients. Circulation 2009, 119, 1661–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobellis, G.; Pistilli, D.; Gucciardo, M.; Leonetti, F.; Miraldi, F.; Brancaccio, G.; Gallo, P.; di Gioia, C.R.T. Adiponectin Expression in Human Epicardial Adipose Tissue In Vivo is Lower in Patients with Coronary Artery Disease. Cytokine 2005, 29, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef]
- Nattel, S.; Burstein, B.; Dobrev, D. Atrial Remodeling and Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2008, 1, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Gabbiani, G. The Myofibroblast in Wound Healing and Fibrocontractive Diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Fitzgibbons, T.; Czech, M. Epicardial and Perivascular Adipose Tissues and Their Influence on Cardiovascular Disease: Basic Mechanisms and Clinical Associations. J. Am. Heart Assoc. 2014, 3, e000582. [Google Scholar] [CrossRef] [Green Version]
- Ouwens, D.; Sell, H.; Greulich, S.; Eckel, J. The Role of Epicardial and Perivascular Adipose Tissue in the Pathophysiology of Cardiovascular Disease. J. Cell. Mol. Med. 2010, 14, 2223–2234. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, M.; Eschholz, N.; Herzberg, S.; Jerchel, I.; Leifheit-Nestler, M.; Czepluch, F.; Chalikias, G.; Konstantinides, S.; Schäfer, K. Leptin-Dependent and Leptin-Independent Paracrine Effects of Perivascular Adipose Tissue on Neointima Formation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 980–987. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.B.; Mori, J.; McLean, B.A.; Basu, R.; Das, S.K.; Ramprasath, T.; Parajuli, N.; Penninger, J.M.; Grant, M.B.; Lopaschuk, G.D.; et al. ACE2 Deficiency Worsens Epicardial Adipose Tissue Inflammation and Cardiac Dysfunction in Response to Diet-Induced Obesity. Diabetes 2016, 65, 85–95. [Google Scholar] [CrossRef] [Green Version]
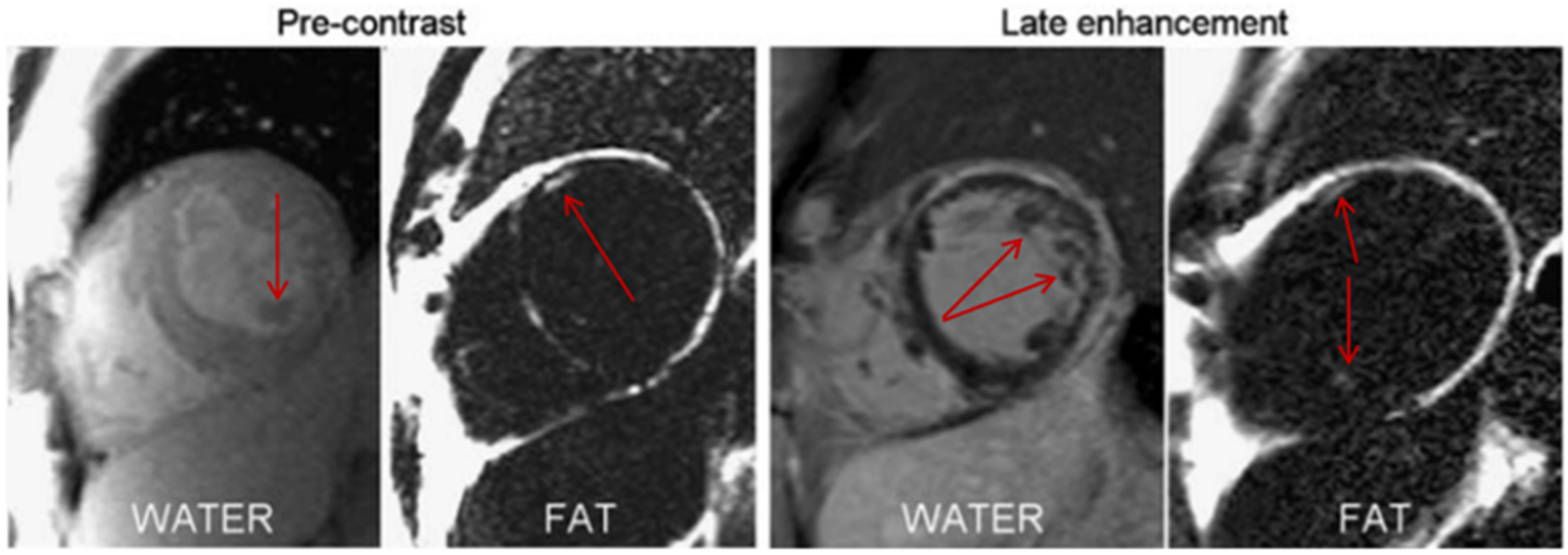
- Ng, A.C.T.; Strudwick, M.; van der Geest, R.J.; Ng, A.C.C.; Gillinder, L.; Goo, S.Y.; Cowin, G.; Delgado, V.; Wang, W.Y.S.; Bax, J.J. Impact of Epicardial Adipose Tissue, Left Ventricular Myocardial Fat Content, and Interstitial Fibrosis on Myocardial Contractile Function. Circ. Cardiovasc. Imaging 2018, 11, e007372. [Google Scholar] [CrossRef] [Green Version]
- Litwin, S. Normal Weight Obesity. Circ. Cardiovasc. Imaging 2012, 5, 286–288. [Google Scholar] [CrossRef] [Green Version]
- Kellman, P.; Hernando, D.; Arai, A. Myocardial Fat Imaging. Curr. Cardiovasc. Imaging Rep. 2010, 3, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Wang, H.; Chen, J.; Zhao, L. Epicardial Adipose Tissue and Atrial Fibrillation: Possible Mechanisms, Potential Therapies, and Future Directions. Pacing Clin. Electrophysiol. 2019, 43, 133–145. [Google Scholar] [CrossRef]
- Santiago-Fernández, C.; Pérez-Belmonte, L.; Millán-Gómez, M.; Moreno-Santos, I.; Carrasco-Chinchilla, F.; Ruiz-Salas, A.; Morcillo-Hidalgo, L.; Melero, J.M.; Garrido-Sánchez, L.; Jiménez-Navarro, M. Overexpression of Scavenger Receptor and Infiltration of Macrophage in Epicardial Adipose Tissue of Patients with Ischemic Heart Disease and Diabetes. J. Transl. Med. 2019, 17, 95. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ Effector T Cells Contribute to Macrophage Recruitment and Adipose Tissue Inflammation in Obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef]
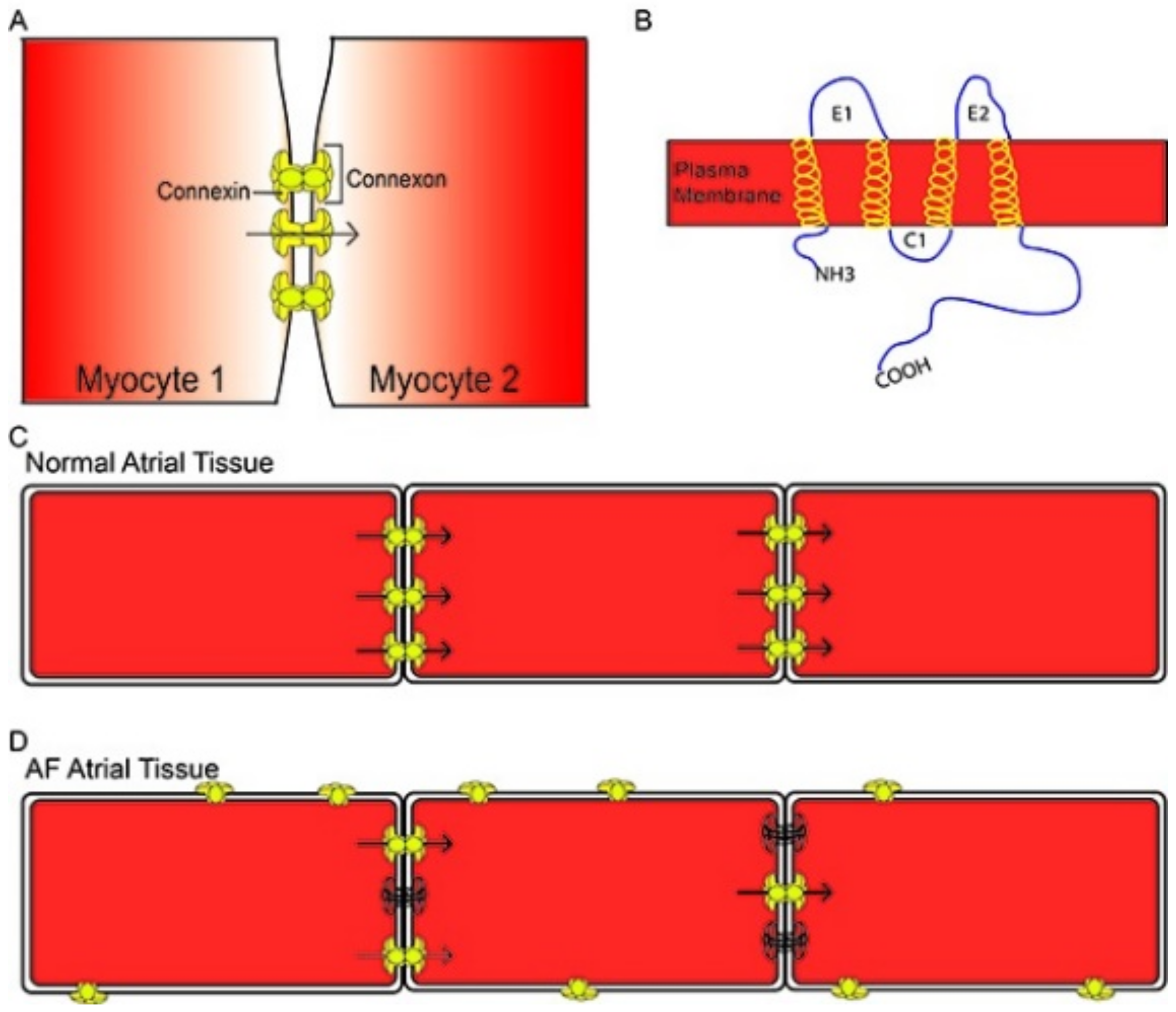
- Jongsma, H.; Wilders, R. Gap Junctions in Cardiovascular Disease. Circ. Res. 2000, 86, 1193–1197. [Google Scholar] [CrossRef]
- Gros, D.; Jongsma, H. Connexins in Mammalian Heart Function. BioEssays 1996, 18, 719–730. [Google Scholar] [CrossRef]
- Asghar, O.; Alam, U.; Hayat, S.A.; Aghamohammadzadeh, R.; Heagerty, A.M.; Malik, R.A. Obesity, Diabetes and Atrial Fibrillation; Epidemiology, Mechanisms and Interventions. Curr. Cardiol. Rev. 2012, 8, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Veasey, R.; Sugihara, C.; Sandhu, K.; Dhillon, G.; Freemantle, N.; Furniss, S.S.; Sulke, A.N. The Natural History of Atrial Fibrillation in Patients with Permanent Pacemakers: Is Atrial Fibrillation a Progressive Disease? J. Interv. Card. Electrophysiol. 2015, 44, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, M.; Pathak, R.; Meredith, M.; Mehta, A.; Elliott, A.; Mahajan, R.; Twomey, D.; Gallagher, C.; Hendriks, J.M.L.; Linz, D.; et al. PREVEntion and regReSsive Effect of Weight-loss and Risk Factor Modification on Atrial Fibrillation: The REVERSE-AF Study. Europace 2018, 20, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; ESC Scientific Document Group; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.M.; Donahue, J.K. Connexin Remodeling Contributes to Atrial Fibrillation. J. Atr. Fibrillation 2013, 6, 839. [Google Scholar] [CrossRef]
- Suffee, N.; Moore-Morris, T.; Jagla, B.; Mougenot, N.; Dilanian, G.; Berthet, M.; Proukhnitzky, J.; Le Prince, P.; Tregouet, D.A.; Pucéat, M.; et al. Reactivation of the Epicardium at the Origin of Myocardial Fibro-Fatty Infiltration During the Atrial Cardiomyopathy. Circ. Res. 2020, 126, 1330–1342. [Google Scholar] [CrossRef]
- Denham, N.C.; Pearman, C.M.; Caldwell, J.L.; Madders, G.W.P.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front. Physiol. 2018, 9, 1380. [Google Scholar] [CrossRef] [Green Version]
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef]
- Anumonwo, J.M.B.; Herron, T. Fatty Infiltration of the Myocardium and Arrhythmogenesis: Potential Cellular and Molecular Mechanisms. Front. Physiol. 2018, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Thanassoulis, G.; Massaro, J.M.; O’Donnell, C.J.; Hoffmann, U.; Levy, D.; Ellinor, P.T.; Wang, T.J.; Schnabel, R.B.; Vasan, R.S.; Fox, C.S.; et al. Pericardial Fat is Associated with Prevalent Atrial Fibrillation: The Framingham Heart Study. Circ. Arrhythm. Electrophysiol. 2010, 3, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Al Chekakie, M.; Welles, C.; Metoyer, R.; Ibrahim, A.; Shapira, A.; Cytron, J.; Santucci, P.; Wilber, D.J.; Akar, J.G. Pericardial Fat Is Independently Associated with Human Atrial Fibrillation. J. Am. Coll. Cardiol. 2010, 56, 784–788. [Google Scholar] [CrossRef] [Green Version]
- Batal, O.; Schoenhagen, P.; Shao, M.; Ayyad, A.E.; Van Wagoner, D.R.; Halliburton, S.S.; Tchou, P.J.; Chung, M.K. Left Atrial Epicardial Adiposity and Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2010, 3, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Maeda, M.; Oba, K.; Yamaguchi, S.; Arasaki, O.; Sata, M.; Masuzaki, H.; Shimabukuro, M. Usefulness of Epicardial Adipose Tissue Volume to Predict Recurrent Atrial Fibrillation After Radiofrequency Catheter Ablation. Am. J. Cardiol. 2018, 122, 1694–1700. [Google Scholar] [CrossRef]
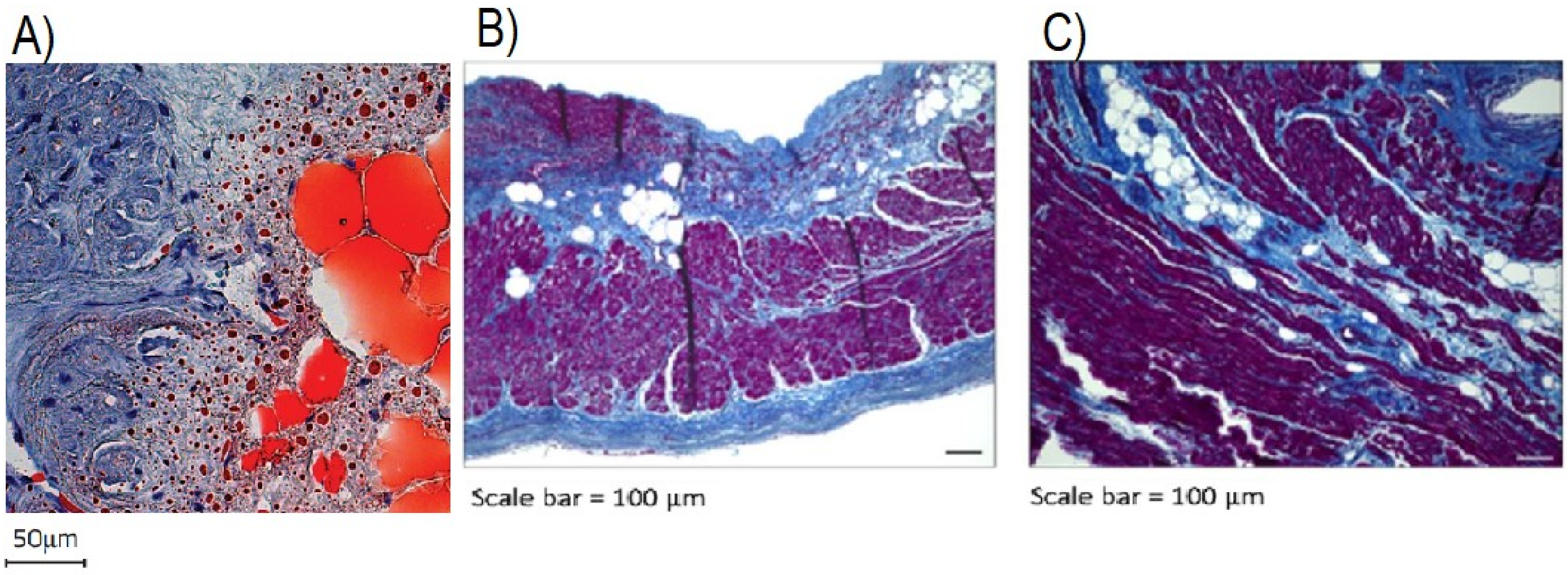
- Haemers, P.; Hamdi, H.; Guedj, K.; Suffee, N.; Farahmand, P.; Popovic, N.; Claus, P.; LePrince, P.; Nicoletti, A.; Jalife, J.; et al. Atrial Fibrillation is Associated with the Fibrotic Remodelling of Adipose Tissue in the Subepicardium of Human and Sheep Atria. Eur. Heart J. 2015, 38, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Fareh, S.; Leung, T.K.; Nattel, S. Promotion of Atrial Fibrillation by Heart Failure in Dogs: Atrial Remodeling of a Different Sort. Circulation 1999, 100, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Abed, H.; Samuel, C.; Lau, D.; Kelly, D.; Royce, S.; Alasady, M.; Mahajan, R.; Kuklik, P.; Zhang, Y.; Brooks, A.G.; et al. Obesity Results in Progressive Atrial Structural and Electrical Remodeling: Implications for Atrial Fibrillation. Heart Rhythm 2013, 10, 90–100. [Google Scholar] [CrossRef]
- Nalliah, C.J.; Bell, J.R.; Raaijmakers, A.J.A.; Waddell, H.M.; Wells, S.P.; Bernasochi, G.B.; Montgomery, M.K.; Binny, S.; Watts, T.; Joshi, S.B.; et al. Epicardial Adipose Tissue Accumulation Confers Atrial Conduction Abnormality. J. Am. Coll. Cardiol. 2020, 76, 1197–1211. [Google Scholar] [CrossRef]
- Abe, I.; Teshima, Y.; Kondo, H.; Kaku, H.; Kira, S.; Ikebe, Y.; Saito, S.; Fukui, A.; Shinohara, T.; Yufu, K.; et al. Association of Fibrotic Remodeling and Cytokines/chemokines Content in Epicardial Adipose Tissue with Atrial Myocardial Fibrosis in Patients with Atrial Fibrillation. Heart Rhythm 2018, 15, 1717–1727. [Google Scholar] [CrossRef]
- Venteclef, N.; Guglielmi, V.; Balse, E.; Gaborit, B.; Cotillard, A.; Atassi, F.; Amour, J.; LePrince, P.; Dutour, A.; Clément, K.; et al. Human Epicardial Adipose Tissue Induces Fibrosis of the Atrial Myocardium through the Secretion of Adipo-fibrokines. Eur. Heart J. 2014, 36, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Min, J.; Jia, L.; Xi, W.; Gao, Y.; Diao, Z.; Zhang, P.; Wang, S.; Yang, J.; Wang, L.; et al. Human Epicardial Adipose Tissue Activin A Expression Predicts Occurrence of Postoperative Atrial Fibrillation in Patients Receiving Cardiac Surgery. Heart Lung Circ. 2019, 28, 1697–1705. [Google Scholar] [CrossRef]
- Kato, T.; Iwasaki, Y.; Nattel, S. Connexins and Atrial Fibrillation. Circulation 2012, 125, 203–206. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liu, T.; Ng, C.; Li, G. Diabetes Mellitus and Atrial Remodeling: Mechanisms and Potential Upstream Therapies. Cardiovasc. Ther. 2014, 32, 233–241. [Google Scholar] [CrossRef]
- Latchamsetty, R.; Morady, F. Complex Fractionated Atrial Electrograms. Circ. Arrhythm. Electrophysiol. 2011, 4, 117–118. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, H.; Yamabe, H.; Enomoto, K.; Koyama, J.; Morihisa, K.; Hoshiyama, T.; Matsui, K.; Ogawa, H. Importance of Pericardial Fat in the Formation of Complex Fractionated Atrial Electrogram Region in Atrial Fibrillation. Int. J. Cardiol. 2014, 174, 557–564. [Google Scholar] [CrossRef]
- Lin, Y.K.; Chen, Y.J.; Chen, S.A. Potential Atrial Arrhythmogenicity of Adipocytes: Implications for the Genesis of Atrial Fibrillation. Med. Hypotheses 2010, 74, 1026–1029. [Google Scholar] [CrossRef]
- Gawałko, M.; Saljic, A.; Li, N.; Abu-Taha, I.; Jespersen, T.; Linz, D.; Nattel, S.; Heijman, J.; Fender, A.; Dobrev, D. Adiposity-associated atrial fibrillation: Molecular determinants, mechanisms and clinical significance. Cardiovasc. Res. 2022, cvac093, Epub ahead of print. [Google Scholar] [CrossRef]
- Martinez-Mateu, L.; Saiz, J.; Aromolaran, A.S. Differential Modulation of IK and ICa,L Channels in High-Fat Diet-Induced Obese Guinea Pig Atria. Front. Physiol. 2019, 10, 1212. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shen, H.; Min, J.; Gao, Y.; Liu, K.; Xi, W.; Yang, J.; Yin, L.; Xu, J.; Xiao, J.; et al. YKL-40 is Highly Expressed in the Epicardial Adipose Tissue of Patients with Atrial Fibrillation and Associated with Atrial Fibrosis. J. Transl. Med. 2018, 16, 229. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.R.; Erikstrup, C.; Johansen, J.S.; Fischer, C.P.; Plomgaard, P.; Krogh-Madsen, R.; Taudorf, S.; Lindegaard, B.; Pedersen, B.K. Plasma YKL-40: A BMI-Independent Marker of Type 2 Diabetes. Diabetes 2008, 57, 3078–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Scott, L., Jr.; Fender, A.C.; Saljic, A.; Li, L.; Chen, X.; Wang, X.; Linz, D.; Lang, J.; Hohl, M.; Twomey, D.; et al. NLRP3 inflammasome is a key driver of obesity-induced atrial arrhythmias. Cardiovasc. Res. 2021, 117, 1746–1759. [Google Scholar] [CrossRef]
- Lin, Y.K.; Chen, Y.C.; Chen, J.H.; Chen, S.A.; Chen, Y.J. Adipocytes Modulate the Electrophysiology of Atrial Myocytes: Implications in Obesity-induced Atrial Fibrillation. Basic Res. Cardiol. 2012, 107, 293. [Google Scholar] [CrossRef]
- Kang, J.X.; Xiao, Y.F.; Leaf, A. Free, Long-chain, Polyunsaturated Fatty Acids Reduce Membrane Electrical Excitability in Neonatal Rat Cardiac Myocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 3997–4001. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, R.; Nelson, A.; Pathak, R.K.; Middeldorp, M.E.; Wong, C.X.; Twomey, D.J.; Carbone, A.; Teo, K.; Agbaedeng, T.; Linz, D.; et al. Electroanatomical Remodeling of the Atria in Obesity. JACC Clin. Electrophysiol. 2018, 4, 1529–1540. [Google Scholar] [CrossRef]
- Lin, Y.K.; Chen, Y.C.; Huang, J.H.; Lin, Y.J.; Huang, S.S.; Chen, S.A.; Chen, Y.J. Leptin Modulates Electrophysiological Characteristics and Isoproterenol-induced Arrhythmogenesis in Atrial Myocytes. J. Biomed. Sci. 2013, 20, 94. [Google Scholar] [CrossRef] [Green Version]
- Australian Institute of Health and Welfare. Cardiovascular Disease, How Many Australians Have Cardiovascular Disease? 2020. Available online: https://www.aihw.gov.au/reports/heart-stroke-vascular-diseases/cardiovascular-health-compendium/contents/how-many-australians-have-cardiovascular-disease (accessed on 21 June 2021).
- Haffner, S.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from Coronary Heart Disease in Subjects with Type 2 Diabetes and in Nondiabetic Subjects with and without Prior Myocardial Infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of Cardiovascular Disease in Type 2 Diabetes: A Systematic Literature Review of Scientific Evidence from Across the World in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.A.; Paneni, F.; Cosentino, F.; Creager, M.A. Diabetes and Vascular Disease: Pathophysiology, Clinical Consequences, and Medical Therapy: Part II. Eur. Heart J. 2013, 34, 2444–2452. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Theroux, P. Pathophysiology of Coronary Artery Disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef] [Green Version]
- Dokken, B.B. The Pathophysiology of Cardiovascular Disease and Diabetes: Beyond Blood Pressure and Lipids. Diabetes Spectr. 2008, 21, 160–165. [Google Scholar] [CrossRef] [Green Version]
- Aronson, D.; Edelman, E. Coronary Artery Disease and Diabetes Mellitus. Cardiol. Clin. 2014, 32, 439–455. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro, M.; Hirata, Y.; Tabata, M.; Dagvasumberel, M.; Sato, H.; Kurobe, H.; Fukuda, D.; Soeki, T.; Kitagawa, T.; Takanashi, S.; et al. Epicardial adipose tissue volume and adipocytokine imbalance are strongly linked to human coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1077–1084. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Fu, J. Novel Insights into the NLRP 3 Inflammasome in Atherosclerosis. J. Am. Heart Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [Green Version]
- Antonopoulos, A.; Tousoulis, D. The Molecular Mechanisms of Obesity Paradox. Cardiovasc. Res. 2017, 113, 1074–1086. [Google Scholar] [CrossRef]
- Kissebah, A.; Krakower, G. Regional Adiposity and Morbidity. Physiol. Rev. 1994, 74, 761–811. [Google Scholar] [CrossRef]
- Moreno-Santos, I.; Pérez-Belmonte, L.M.; Macías-González, M.; Mataró, M.J.; Castellano, D.; López Garrido, M.; Porras Martín, C.; Sánchez Fernández, P.L.; Gómez Doblas, J.J.; Cardona, F.; et al. Type 2 Diabetes is Associated with Decreased PGC1α Expression in Epicardial Adipose Tissue of Patients with Coronary Artery Disease. J. Transl. Med. 2016, 14, 243. [Google Scholar] [CrossRef] [Green Version]
- Chechi, K.; Blanchard, P.; Mathieu, P.; Deshaies, Y.; Richard, D. Brown Fat Like Gene Expression in the Epicardial Fat Depot Correlates with Circulating HDL-cholesterol and Triglycerides in Patients with Coronary Artery Disease. Int. J. Cardiol. 2013, 167, 2264–2270. [Google Scholar] [CrossRef]
- Singh, S.P.; McClung, J.A.; Thompson, E.; Glick, Y.; Greenberg, M.; Acosta-Baez, G.; Edris, B.; Shapiro, J.I.; Abraham, N.G. Cardioprotective Heme Oxygenase-1-PGC1α Signaling in Epicardial Fat Attenuates Cardiovascular Risk in Humans as in Obese Mice. Obesity 2019, 27, 1634–1643. [Google Scholar] [CrossRef]
- Christensen, R.H.; von Scholten, B.J.; Hansen, C.S.; Heywood, S.E.; Rosenmeier, J.B.; Andersen, U.B.; Hovind, P.; Reinhard, H.; Parving, H.H.; Pedersen, B.K.; et al. Epicardial, Pericardial and Total Cardiac Fat and Cardiovascular Disease in Type 2 Diabetic Patients with Elevated Urinary Albumin Excretion Rate. Eur. J. Prev. Cardiol. 2017, 24, 1517–1524. [Google Scholar] [CrossRef]
- Christensen, R.H.; von Scholten, B.J.; Hansen, C.S.; Jensen, M.T.; Vilsbøll, T.; Rossing, P.; Jørgensen, P.G. Epicardial Adipose Tissue Predicts Incident Cardiovascular Disease and Mortality in Patients with Type 2 Diabetes. Cardiovasc. Diabetol. 2019, 18, 114. [Google Scholar] [CrossRef] [Green Version]
- Yerramasu, A.; Dey, D.; Venuraju, S.; Anand, D.V.; Atwal, S.; Corder, R.; Berman, D.S.; Lahiri, A. Increased Volume of Epicardial Fat is an Independent Risk Factor for Accelerated Progression of Sub-clinical Coronary Atherosclerosis. Atherosclerosis 2012, 220, 223–230. [Google Scholar] [CrossRef]
- Reinhardt, M.; Cushman, T.R.; Thearle, M.S.; Krakoff, J. Epicardial Adipose Tissue is a Predictor of Decreased Kidney Function and Coronary Artery Calcification in Youth- and Early Adult-Onset Type 2 Diabetes Mellitus. J. Endocrinol. Investig. 2019, 42, 979–986. [Google Scholar] [CrossRef]
- Kim, H.M.; Kim, K.J.; Lee, H.J.; Yu, H.T.; Moon, J.H.; Kang, E.S.; Cha, B.S.; Lee, H.C.; Lee, B.W.; Kim, Y.J. Epicardial Adipose Tissue Thickness is an Indicator for Coronary Artery Stenosis in Asymptomatic Type 2 Diabetic Patients: Its Assessment by Cardiac Magnetic Resonance. Cardiovasc. Diabetol. 2012, 11, 83. [Google Scholar] [CrossRef] [Green Version]
- Gullaksen, S.; Funck, K.L.; Laugesen, E.; Hansen, T.K.; Dey, D.; Poulsen, P.L. Volumes of Coronary Plaque Disease in Relation to Body Mass Index, Waist Circumference, Truncal Fat Mass and Epicardial Adipose Tissue in Patients with Type 2 Diabetes Mellitus and Controls. Diab. Vasc. Dis. Res. 2019, 16, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, E.V.; Marwan, M.; Desai, M.Y.; Mancio, J.; Alashi, A.; Centeno, E.H.; Thomas, S.; Herdman, L.; Kotanidis, C.P.; Thomas, K.E.; et al. Non-invasive Detection of Coronary Inflammation using Computed Tomography and Prediction of Residual Cardiovascular Risk (the CRISP CT study): A Post-hoc Analysis of Prospective Outcome Data. Lancet 2018, 392, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Tordjman, J.; Clément, K.; Scherer, P.E. Fibrosis and Adipose Tissue Dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konwerski, M.; Postuła, M.; Barczuk-Falęcka, M.; Czajkowska, A.; Mróz, A.; Witek, K.; Bakalarski, W.; Gąsecka, A.; Małek, Ł.A.; Mazurek, T. Epicardial Adipose Tissue and Cardiovascular Risk Assessment in Ultra-Marathon Runners: A Pilot Study. Int. J. Environ. Res. Public Health 2021, 18, 3136. [Google Scholar] [CrossRef] [PubMed]
- Pasarica, M.; Gowronska-Kozak, B.; Burk, D.; Remedios, I.; Hymel, D.; Gimble, J.; Ravussin, E.; Bray, G.A.; Smith, S.R. Adipose Tissue Collagen VI in Obesity. J. Clin. Endocrinol. Metab. 2009, 94, 5155–5162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamdar, A.A.; Inamdar, A.C. Heart Failure: Diagnosis, Management and Utilization. J. Clin. Med. 2016, 5, 62. [Google Scholar] [CrossRef]
- Morrissey, R.P.; Czer, L.; Shah, P.K. Chronic Heart Failure. Am. J. Cardiovasc. Drugs 2011, 11, 153–171. [Google Scholar] [CrossRef]
- Gulsin, G.G.; Athithan, L.; McCann, G.P. Diabetic Cardiomyopathy: Prevalence, Determinants and Potential Treatments. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018819834869. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.; Sowers, J. Diabetic Cardiomyopathy. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Poirier, P.; Bogaty, P.; Garneau, C.; Marois, L.; Dumesnil, J.G. Diastolic Dysfunction in Normotensive Men with Well-Controlled Type 2 Diabetes: Importance of Maneuvers in Echocardiographic Screening for Preclinical Diabetic Cardiomyopathy. Diabetes Care 2001, 24, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Nichols, G.A.; Gullion, C.M.; Koro, C.E.; Ephross, S.A.; Brown, J.B. The Incidence of Congestive Heart Failure in Type 2 Diabetes: An update. Diabetes Care 2004, 27, 1879–1884. [Google Scholar] [CrossRef] [Green Version]
- Lehrke, M.; Marx, N. Diabetes Mellitus and Heart Failure. Am. J. Cardiol. 2017, 120, S37–S47. [Google Scholar] [CrossRef] [Green Version]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.W.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Robert, C.; Turner, R.C.; Holma, R.R.; et al. Association of Glycaemia with Macrovascular and Microvascular Complications of Type 2 Diabetes (UKPDS 35): Prospective Observational Study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Van Melle, J.P.; Bot, M.; de Jonge, P.; de Boer, R.A.; van Veldhuisen, D.J.; Whooley, M.A. Diabetes, Glycemic Control, and New-Onset Heart Failure in Patients with Stable Coronary Artery Disease: Data from the Heart and Soul Study. Diabetes Care 2010, 33, 2084–2089. [Google Scholar] [CrossRef] [Green Version]
- Matloch, Z.; Kotulák, T.; Haluzík, M. The Role of Epicardial Adipose Tissue in Heart Disease. Physiol. Res. 2016, 65, 23–32. [Google Scholar] [CrossRef]
- Westermeier, F.; Riquelme, J.A.; Pavez, M.; Garrido, V.; Díaz, A.; Verdejo, H.E.; Castro, P.F.; García, L.; Lavandero, S. New Molecular Insights of Insulin in Diabetic Cardiomyopathy. Front. Physiol. 2016, 7, 125. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.Y.; Prins, J.B.; Marwick, T.H. Diabetic Cardiomyopathy: Evidence, Mechanisms, and Therapeutic Implications. Endocrine Rev. 2004, 25, 543–567. [Google Scholar] [CrossRef]
- Dzeshka, M.S.; Lip, G.Y.H.; Snezhitskiy, V.; Shantsila, E. Cardiac Fibrosis in Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 2015, 66, 943–959. [Google Scholar] [CrossRef] [Green Version]
- Parisi, V.; Rengo, G.; Perrone-Filardi, P.; Pagano, G.; Femminella, G.D.; Paolillo, S.; Petraglia, L.; Gambino, G.; Caruso, A.; Grimaldi, M.G.; et al. Increased Epicardial Adipose Tissue Volume Correlates With Cardiac Sympathetic Denervation in Patients With Heart Failure. Circ. Res. 2016, 118, 1244–1253. [Google Scholar] [CrossRef] [Green Version]
- Paolillo, S.; Rengo, G.; Pagano, G.; Pellegrino, T.; Savarese, G.; Femminella, G.D.; Tuccillo, M.; Boemio, A.; Attena, E.; Formisano, R.; et al. Impact of Diabetes on Cardiac Sympathetic Innervation in Patients with Heart Failure: A 123I Meta-iodobenzylguanidine (123I MIBG) Scintigraphic Study. Diabetes Care 2013, 36, 2395–2401. [Google Scholar] [CrossRef] [Green Version]
- Çetin, M.; Kocaman, S.A.; Durakoĝlugil, M.E.; Erdoğan, T.; Ergül, E.; Dogan, S.; Canga, A. Effect of Epicardial Adipose Tissue on Diastolic Functions and Left Atrial Dimension in Untreated Hypertensive Patients with Normal Systolic Function. J. Cardiol. 2013, 61, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Christensen, R.H.; Hansen, C.S.; von Scholten, B.J.; Jensen, M.T.; Pedersen, B.K.; Schnohr, P.; Vilsbøll, T.; Rossing, P.; Jørgensen, P.G. Epicardial and Pericardial Adipose Tissues are Associated with Reduced Diastolic and Systolic Function in Type 2 Diabetes. Diabetes Obes. Metab. 2019, 21, 2006–2011. [Google Scholar] [CrossRef]
- Takahari, K.; Utsunomiya, H.; Itakura, K.; Yamamoto, H.; Nakano, Y. Impact of the Distribution of Epicardial and Visceral Adipose Tissue on Left Ventricular Diastolic Function. Heart Vessel. 2022, 37, 250–261. [Google Scholar] [CrossRef]
- Levelt, E.; Pavlides, M.; Banerjee, R.; Mahmod, M.; Kelly, C.; Sellwood, J.; Ariga, R.; Thomas, S.; Francis, J.; Rodgers, C.; et al. Ectopic and Visceral Fat Deposition in Lean and Obese Patients with Type 2 Diabetes. J. Am. Coll. Cardiol. 2016, 68, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Mytas, D.Z.; Stougiannos, P.N.; Zairis, M.N.; Foussas, S.G.; Pyrgakis, V.N.; Kyriazis, I.A. Diabetic Myocardial Disease: Pathophysiology, Early Diagnosis and Therapeutic Options. J. Diabetes Complicat. 2009, 23, 273–282. [Google Scholar] [CrossRef]
- Isfort, M.; Stevens, S.C.; Schaffer, S.; Jong, C.J.; Wold, L.E. Metabolic Dysfunction in Diabetic Cardiomyopathy. Heart Fail. Rev. 2013, 19, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Belmonte, L.; Moreno-Santos, I.; Gómez-Doblas, J.; García-Pinilla, J.; Morcillo-Hidalgo, L.; Garrido-Sánchez, L.; Santiago-Fernández, C.; Crespo-Leiro, M.G.; Carrasco-Chinchilla, F.; Sánchez-Fernández, P.L.; et al. Expression of Epicardial Adipose Tissue Thermogenic Genes in Patients with Reduced and Preserved Ejection Fraction Heart Failure. Int. J. Med. Sci. 2017, 14, 891–895. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Villasante Fricke, A. Effects of Semaglutide Versus Dulaglutide on Epicardial Fat Thickness in Subjects with Type 2 Diabetes and Obesity. J. Endocr. Soc. 2020, 4, bvz042. [Google Scholar] [CrossRef]
- Yagi, S.; Hirata, Y.; Ise, T.; Kusunose, K.; Yamada, H.; Fukuda, D.; Salim, H.M.; Maimaituxun, G.; Nishio, S.; Takagawa, Y.; et al. Canagliflozin Reduces Epicardial Fat in Patients with Type 2 Diabetes Mellitus. Diabetol. Metab. Syndr. 2017, 9, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziyrek, M.; Kahraman, S.; Ozdemir, E.; Dogan, A. Metformin Monotherapy Significantly Decreases Epicardial Adipose Tissue Thickness in Newly Diagnosed Type 2 Diabetes Patients. Rev. Port. Cardiol. 2019, 38, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Raggi, P.; Davidson, M.; Callister, T.Q.; Welty, F.K.; Bachmann, G.A.; Hecht, H.; Rumberger, J.A. Aggressive versus moderate lipid-lowering therapy in hypercholesterolemic postmenopausal women: Beyond Endorsed Lipid Lowering with EBT Scanning (BELLES). Circulation 2005, 112, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Park, Y.S.; Kim, Y.J.; Lee, I.S.; Kim, J.H.; Lee, J.H.; Choi, S.W.; Jeong, J.O.; Seong, I.W. Effects of statins on the epicardial fat thickness in patients with coronary artery stenosis underwent percutaneous coronary intervention: Comparison of atorvastatin with simvastatin/ezetimibe. J. Cardiovasc. Ultrasound 2010, 18, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, V.; Petraglia, L.; D’Esposito, V.; Cabaro, S.; Rengo, G.; Caruso, A.; Grimaldi, M.G.; Baldascino, F.; De Bellis, A.; Vitale, D.; et al. Statin therapy modulates thickness and inflammatory profile of human epicardial adipose tissue. Int. J. Cardiol. 2019, 274, 326–330. [Google Scholar] [CrossRef] [Green Version]
- Raggi, P.; Gadiyaram, V.; Zhang, C.; Chen, Z.; Lopaschuk, G.; Stillman, A.E. Statins Reduce Epicardial Adipose Tissue Attenuation Independent of Lipid Lowering: A Potential Pleiotropic Effect. J. Am. Heart Assoc. 2019, 8, e013104. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Ramos, D.; Mehta, R.; Aguilar-Salinas, C.A. Fibroblast Growth Factor 21 and Browning of White Adipose Tissue. Front. Physiol. 2019, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.D.; Ashmore, T.; Kotwica, A.O.; Murfitt, S.A.; Fernandez, B.O.; Feelisch, M.; Murray, A.J.; Griffin, J.L. Inorganic Nitrate Promotes the Browning of White Adipose Tissue Through the Nitrate-Nitrite-Nitric Oxide Pathway. Diabetes 2014, 64, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Kleinaki, Z.; Agouridis, A.; Zafeiri, M.; Xanthos, T.; Tsioutis, C. Epicardial Adipose Tissue Deposition in Patients with Diabetes and Renal Impairment: Analysis of the Literature. World J. Diabetes 2020, 11, 33–41. [Google Scholar] [CrossRef]

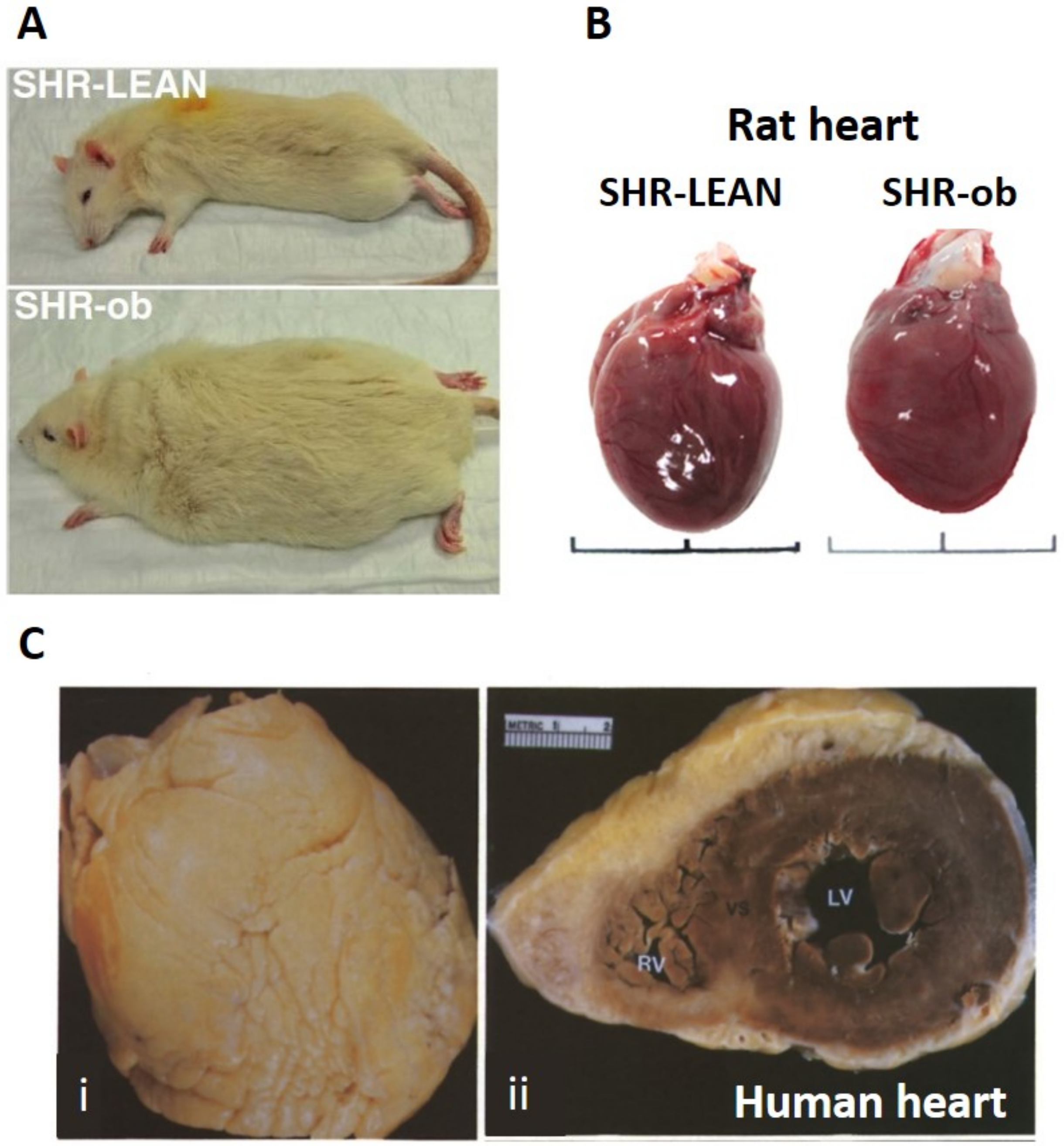





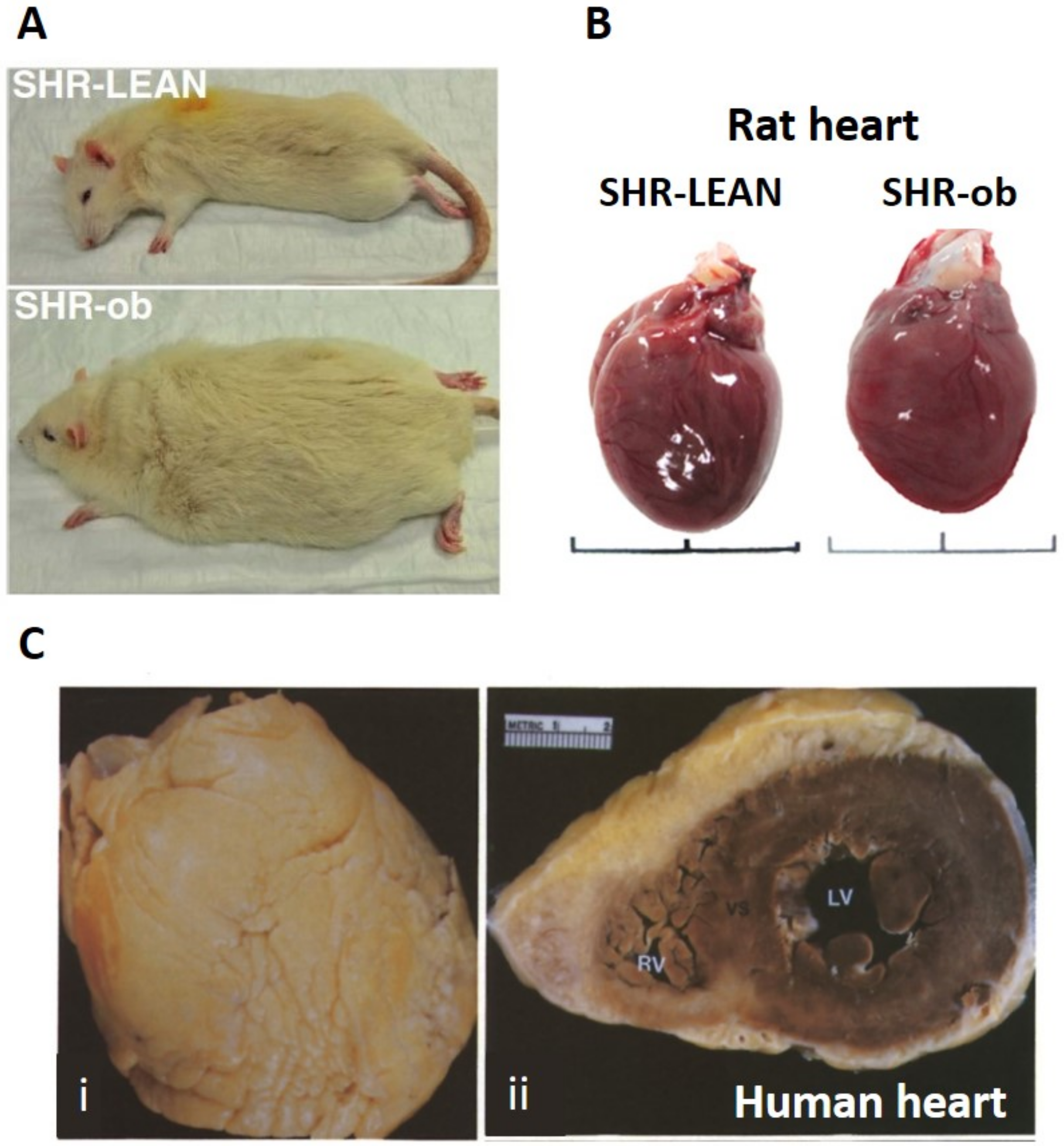
{kind=link}
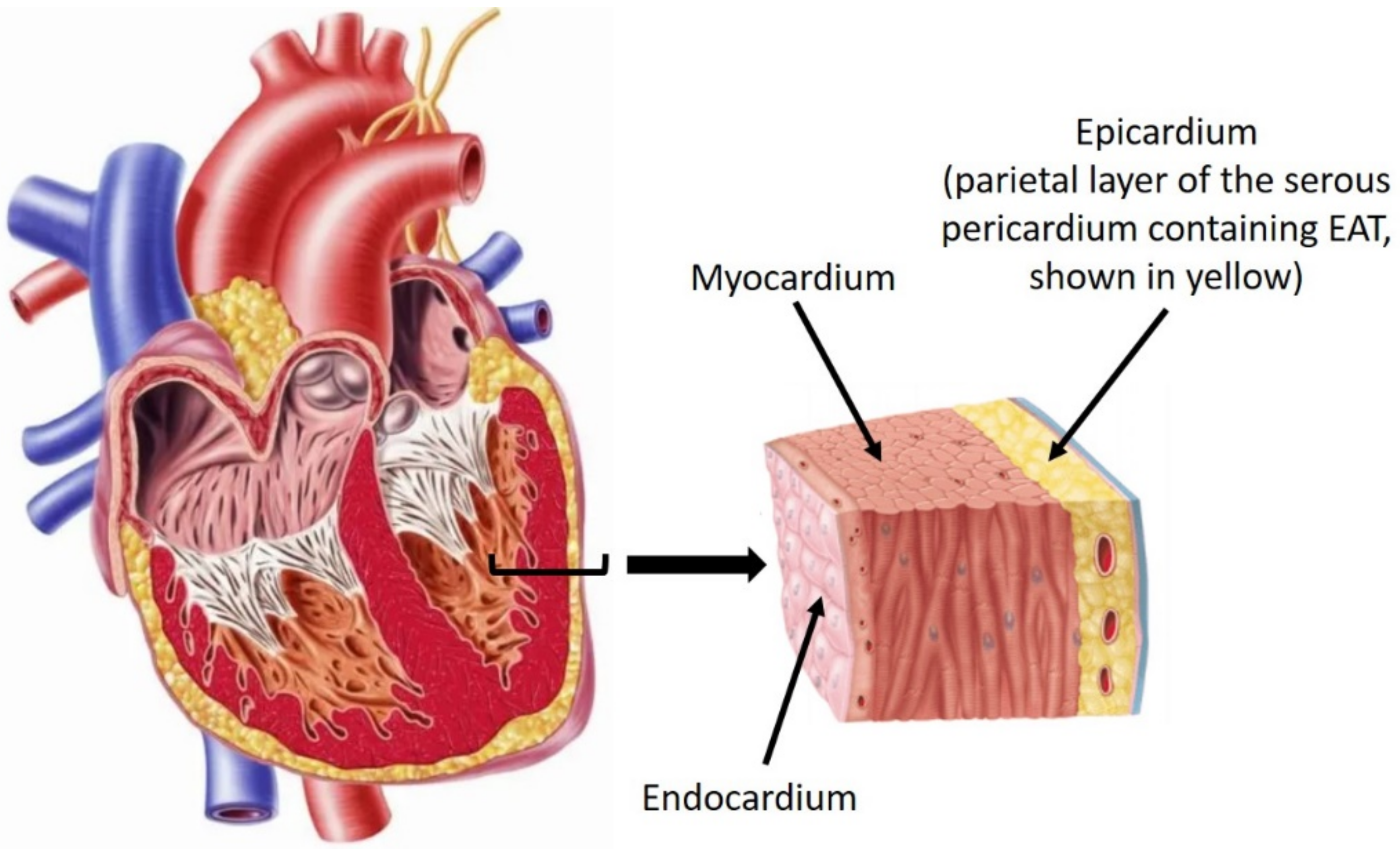
{kind=link}
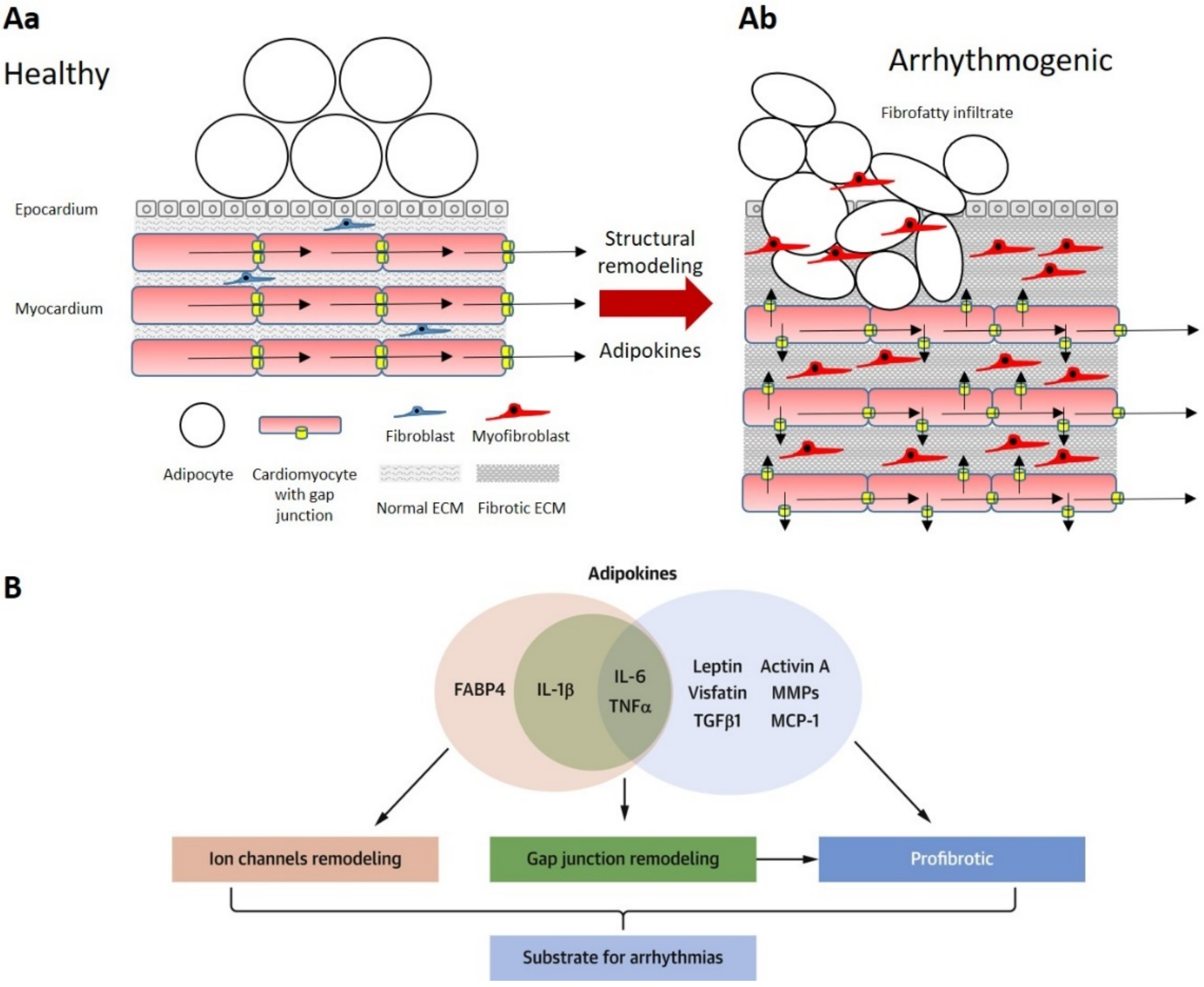{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-Inflammatory, Protective Adipokines | Pro-Inflammatory, Pathologic Adipokines | ||
|---|---|---|---|
| Factor | Effect | Factor | Effect |
| Adiponectin | Vasodilation, prevention of monocyte adhesion to endothelium, promotes local nitric oxide action [40]. | TNF-α, IL-1, IL-6, IL-8 | Released from immune cells and adipocytes. Induce lipolysis, promote apoptosis, inhibit adiponectin secretion, recruit nearby immune cells [25]. |
| Leptin | Endothelium-dependent vasodilation [40]. | Resistin | Released from macrophages and adipocytes. Released in response to TNF-α [25]. |
| Omentin-1 | Inhibits TGF-β and limits fibroblast differentiation and collagen deposition [25]. | Visfatin | Acts as an insulin-mimetic and is a marker of visceral fat. Thought to play a role in vascular inflammation and remodeling [41]. |
| Adrenomedullin | Antioxidant, angiogenic, vasodilatory, and anti-inflammatory properties. Inhibits nuclear factor kappa B (NF-κB) pathway involved in inflammation [25]. | Leptin | Enhance smooth muscle cell proliferation, angiogenesis, platelet aggregation and increased leptin expression on atherosclerotic lesions suggest atherogenic role [42]. |
| UCP1/PGC-1α | Proteins aid in mitochondrial release of heat through consumption of chemical energy–thermogenic capability [18]. | Ang II | Promotes macrophage polarisation to inflammatory phenotype, promoting production of fibrotic cytokines (TGF-β), vasoconstriction [43]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, A.; Sharma, H.; Yuan, D.; Trollope, A.F.; Chilton, L. The Role of Epicardial Adipose Tissue in the Development of Atrial Fibrillation, Coronary Artery Disease and Chronic Heart Failure in the Context of Obesity and Type 2 Diabetes Mellitus: A Narrative Review. J. Cardiovasc. Dev. Dis. 2022, 9, 217. https://doi.org/10.3390/jcdd9070217
Krishnan A, Sharma H, Yuan D, Trollope AF, Chilton L. The Role of Epicardial Adipose Tissue in the Development of Atrial Fibrillation, Coronary Artery Disease and Chronic Heart Failure in the Context of Obesity and Type 2 Diabetes Mellitus: A Narrative Review. Journal of Cardiovascular Development and Disease. 2022; 9(7):217. https://doi.org/10.3390/jcdd9070217
Chicago/Turabian StyleKrishnan, Anirudh, Harman Sharma, Daniel Yuan, Alexandra F. Trollope, and Lisa Chilton. 2022. "The Role of Epicardial Adipose Tissue in the Development of Atrial Fibrillation, Coronary Artery Disease and Chronic Heart Failure in the Context of Obesity and Type 2 Diabetes Mellitus: A Narrative Review" Journal of Cardiovascular Development and Disease 9, no. 7: 217. https://doi.org/10.3390/jcdd9070217
APA StyleKrishnan, A., Sharma, H., Yuan, D., Trollope, A. F., & Chilton, L. (2022). The Role of Epicardial Adipose Tissue in the Development of Atrial Fibrillation, Coronary Artery Disease and Chronic Heart Failure in the Context of Obesity and Type 2 Diabetes Mellitus: A Narrative Review. Journal of Cardiovascular Development and Disease, 9(7), 217. https://doi.org/10.3390/jcdd9070217