
Microscopic and Macroscopic Characterization of Hydrogels Based on Poly(vinyl-alcohol)–Glutaraldehyde Mixtures for Fricke Gel Dosimetry

 , , ,
, , , 
Abstract
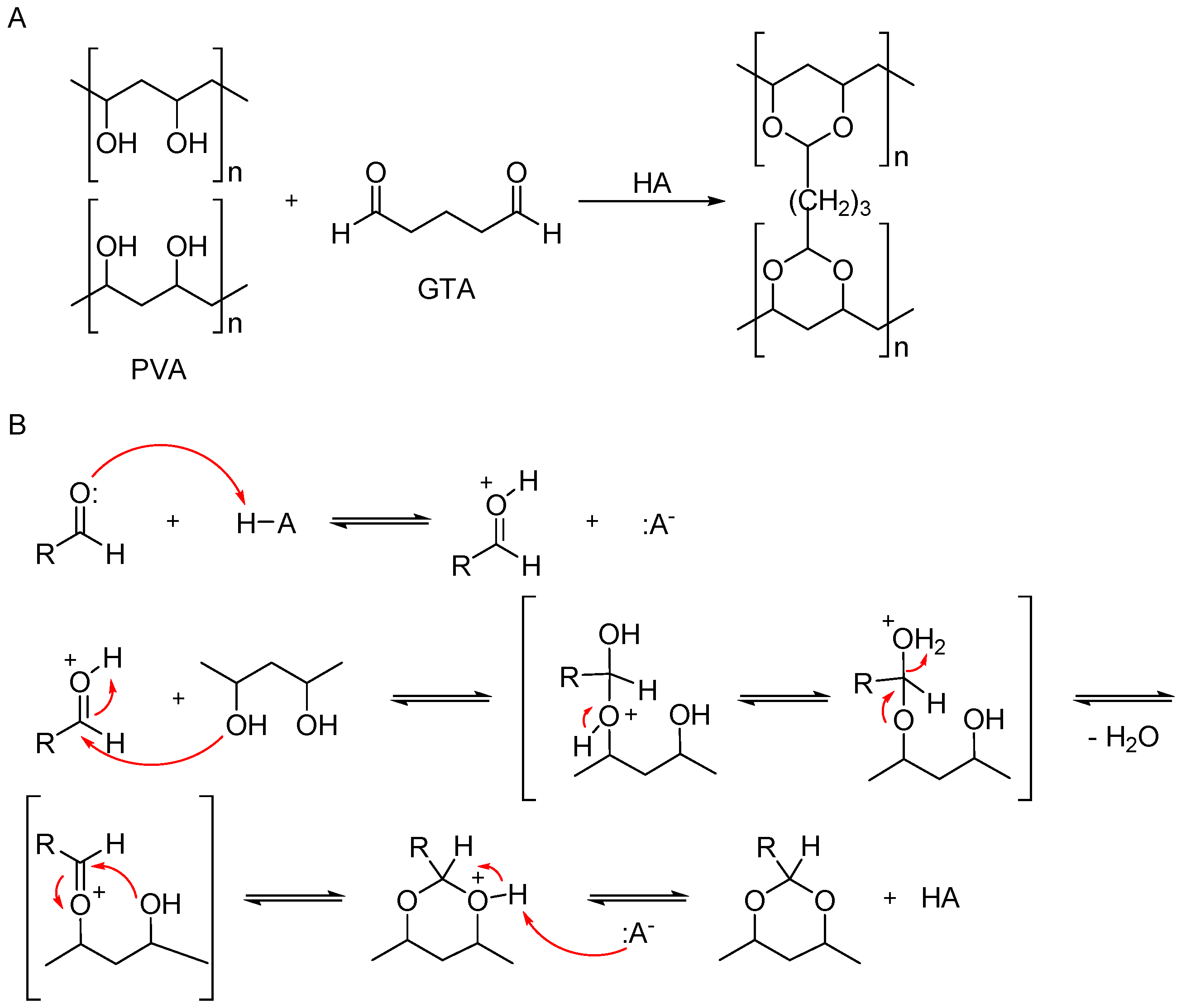
1. Introduction
2. Results and Discussion
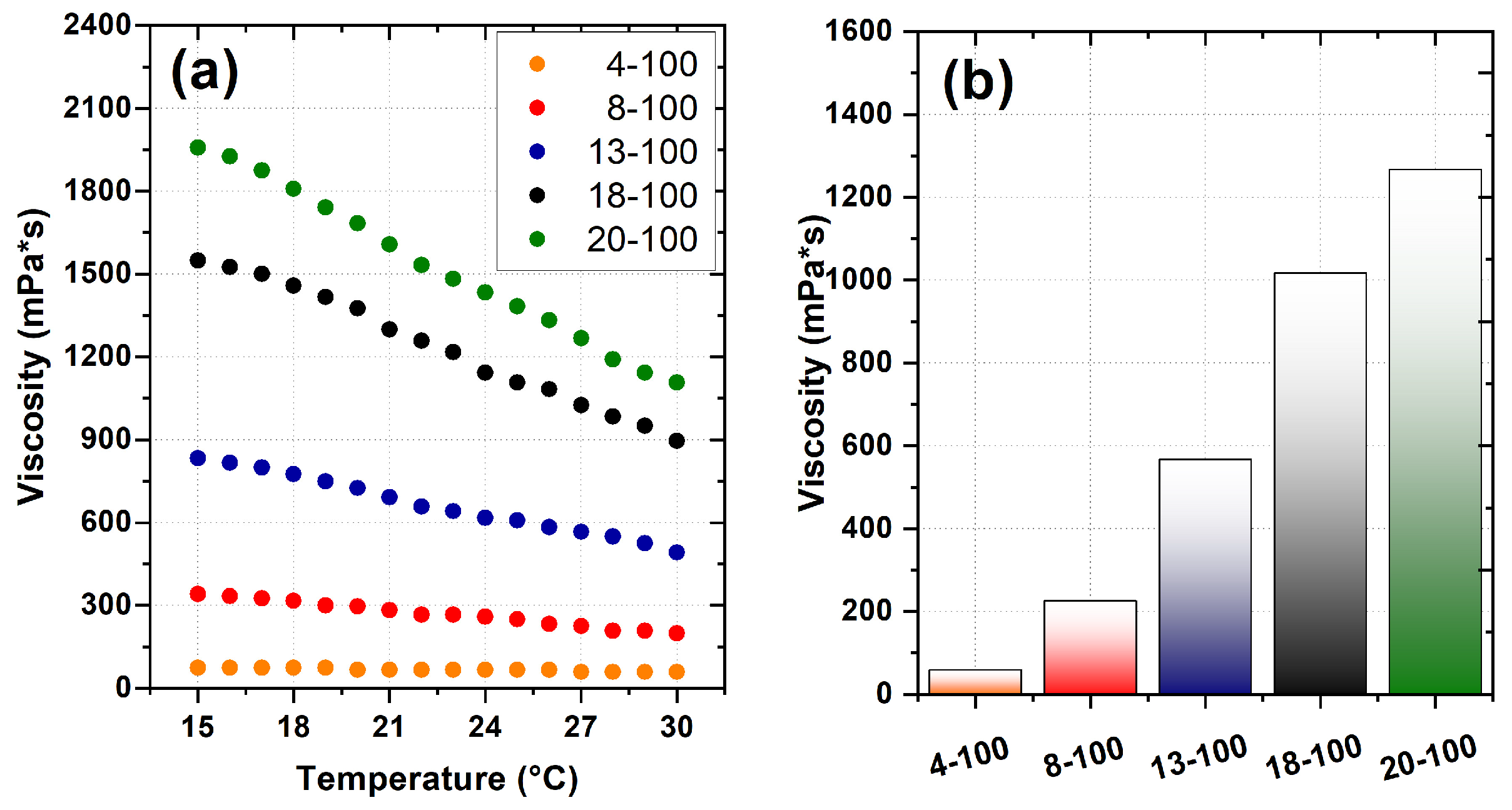
2.1. Sample Preparation and Viscosity Determination
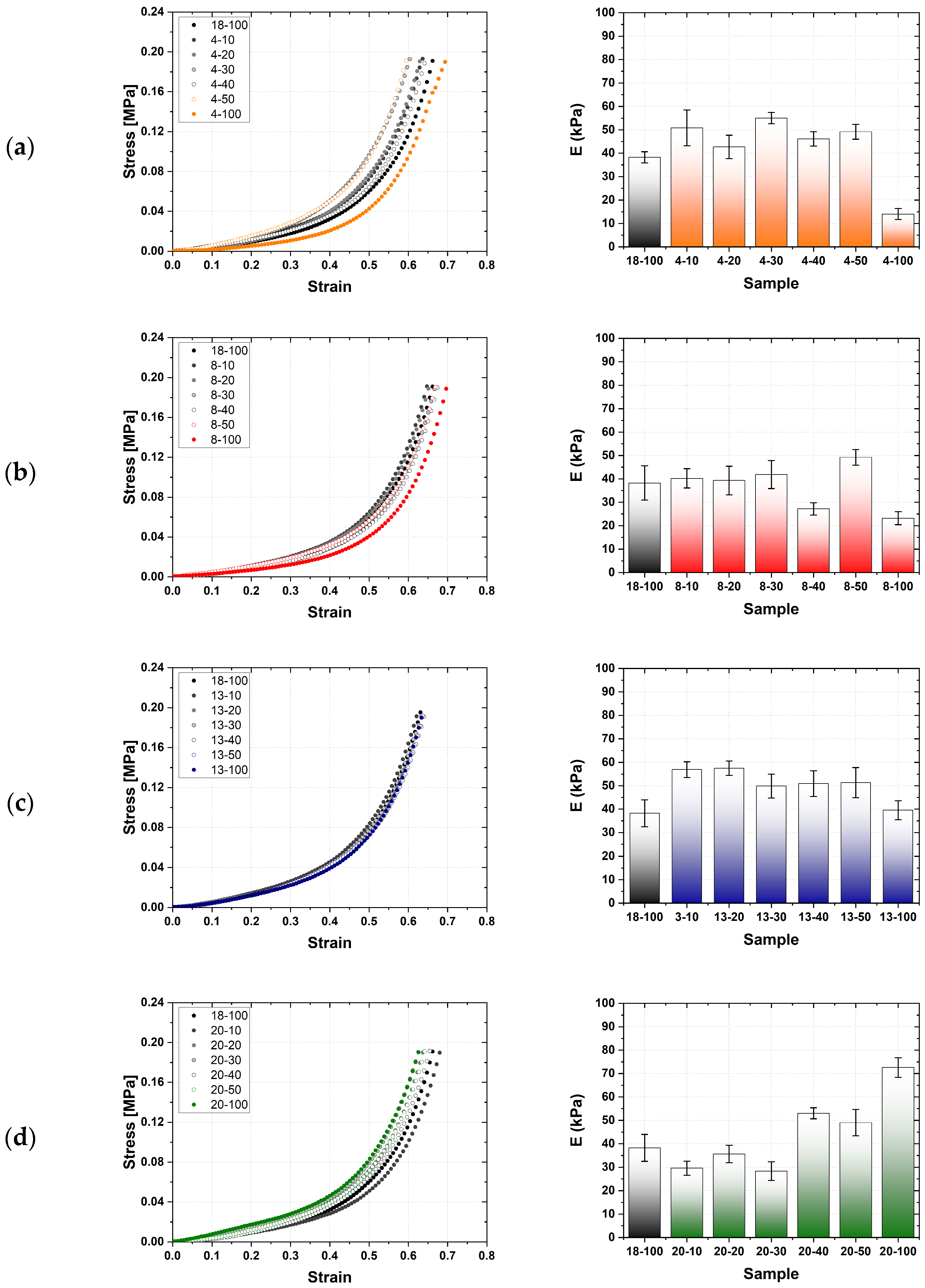
2.2. Mechanical Characterization of Hydrogels
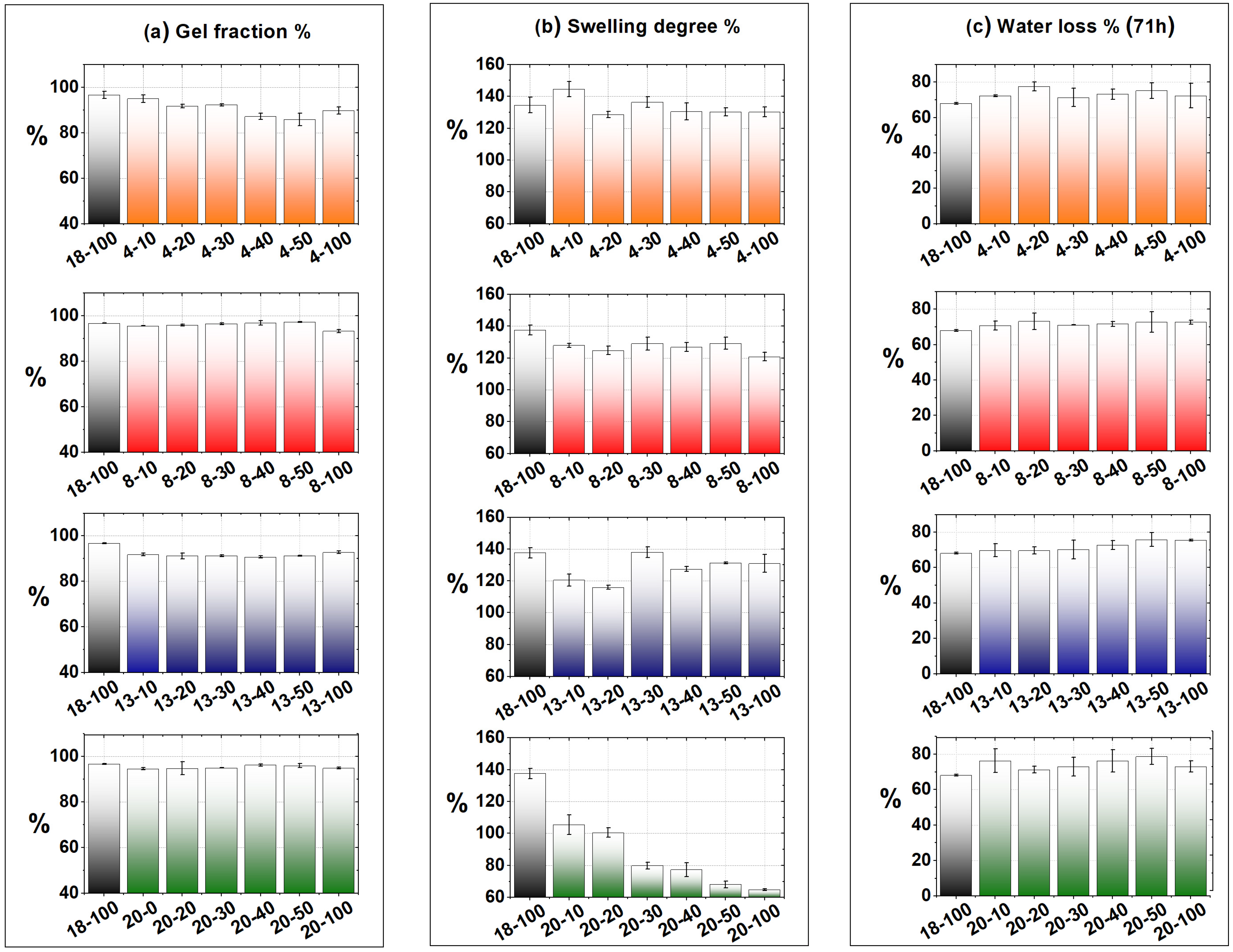
2.3. Physical–Chemical Characterization of the Hydrogels
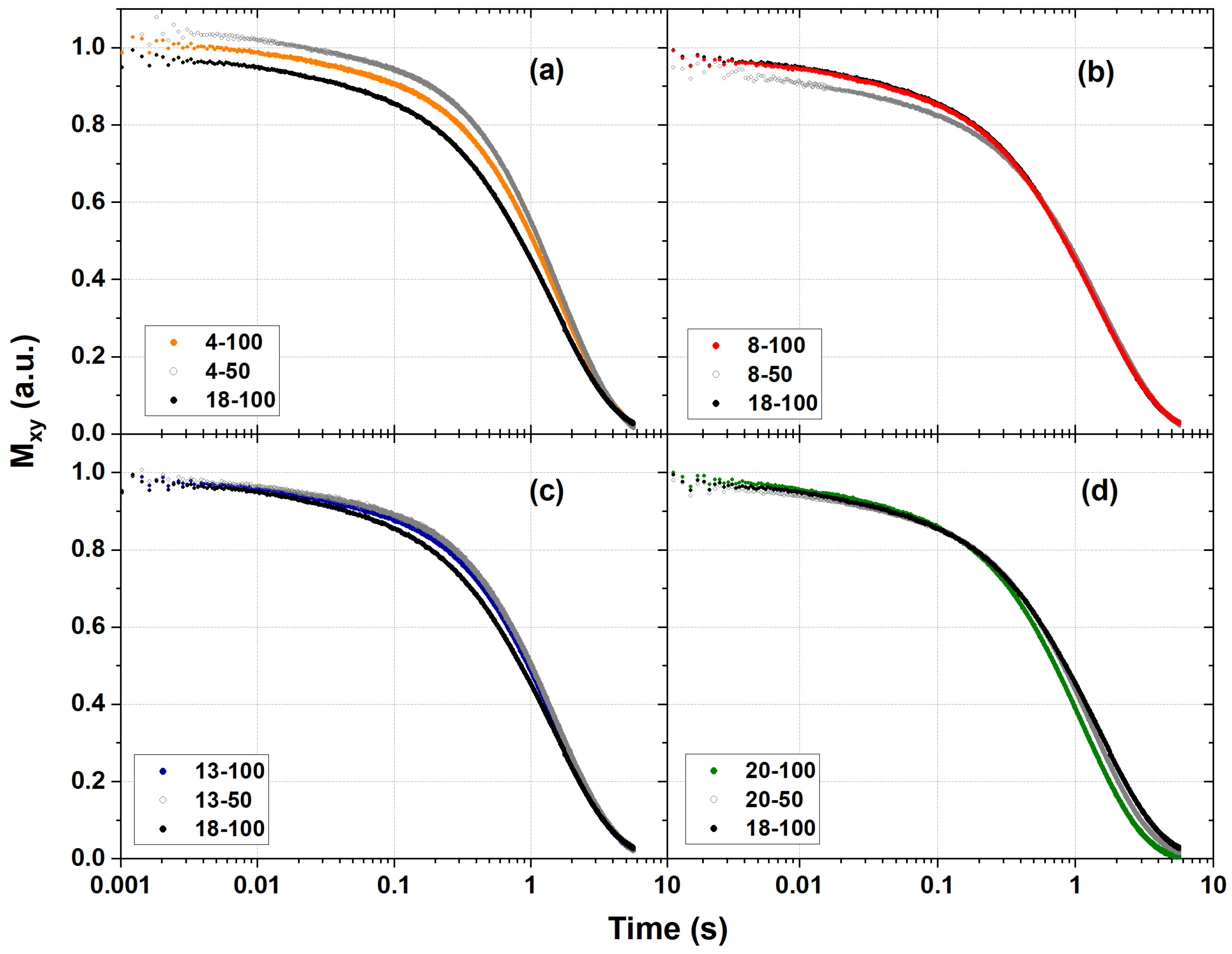
2.4. 1H NMR Relaxometry Measurements
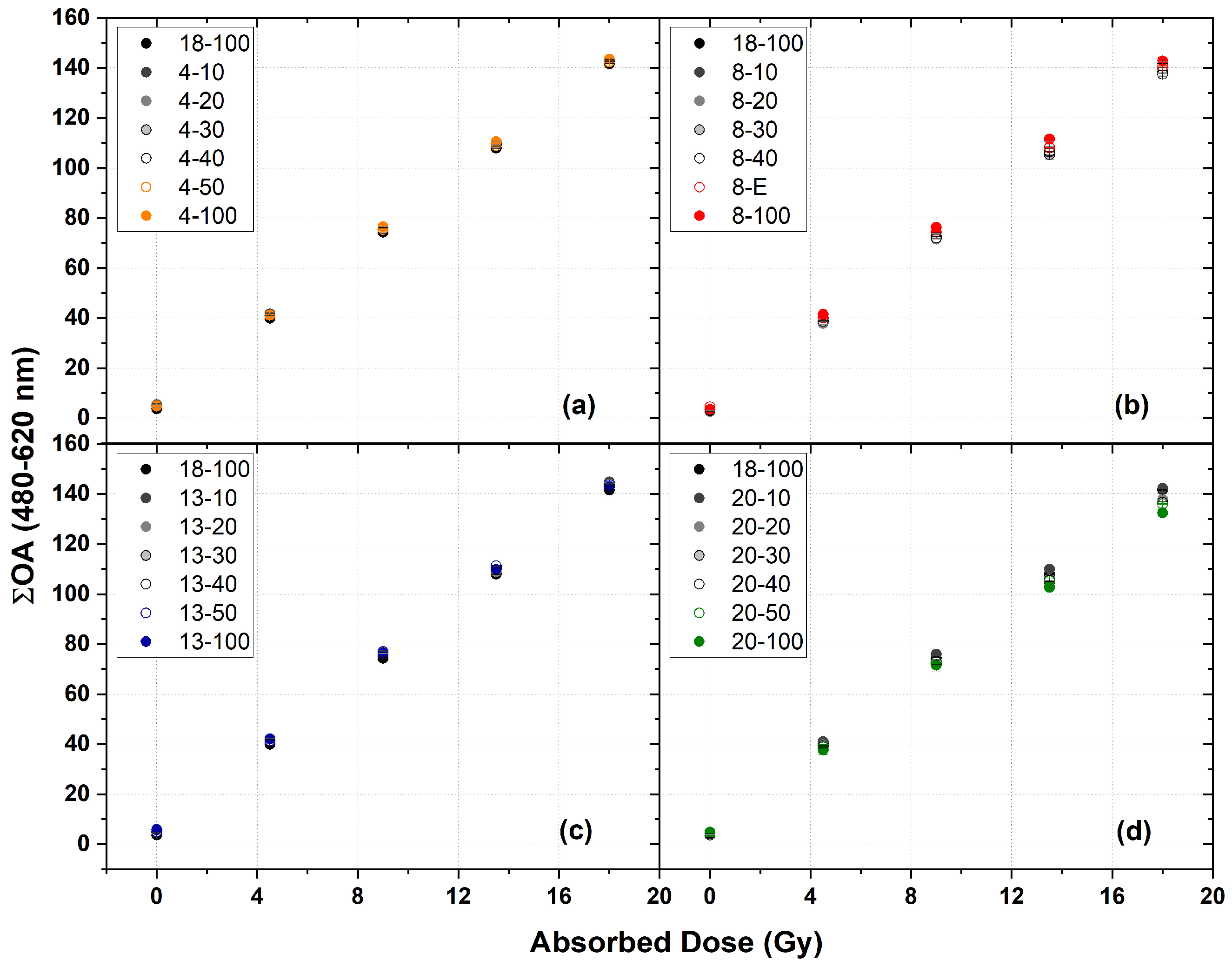
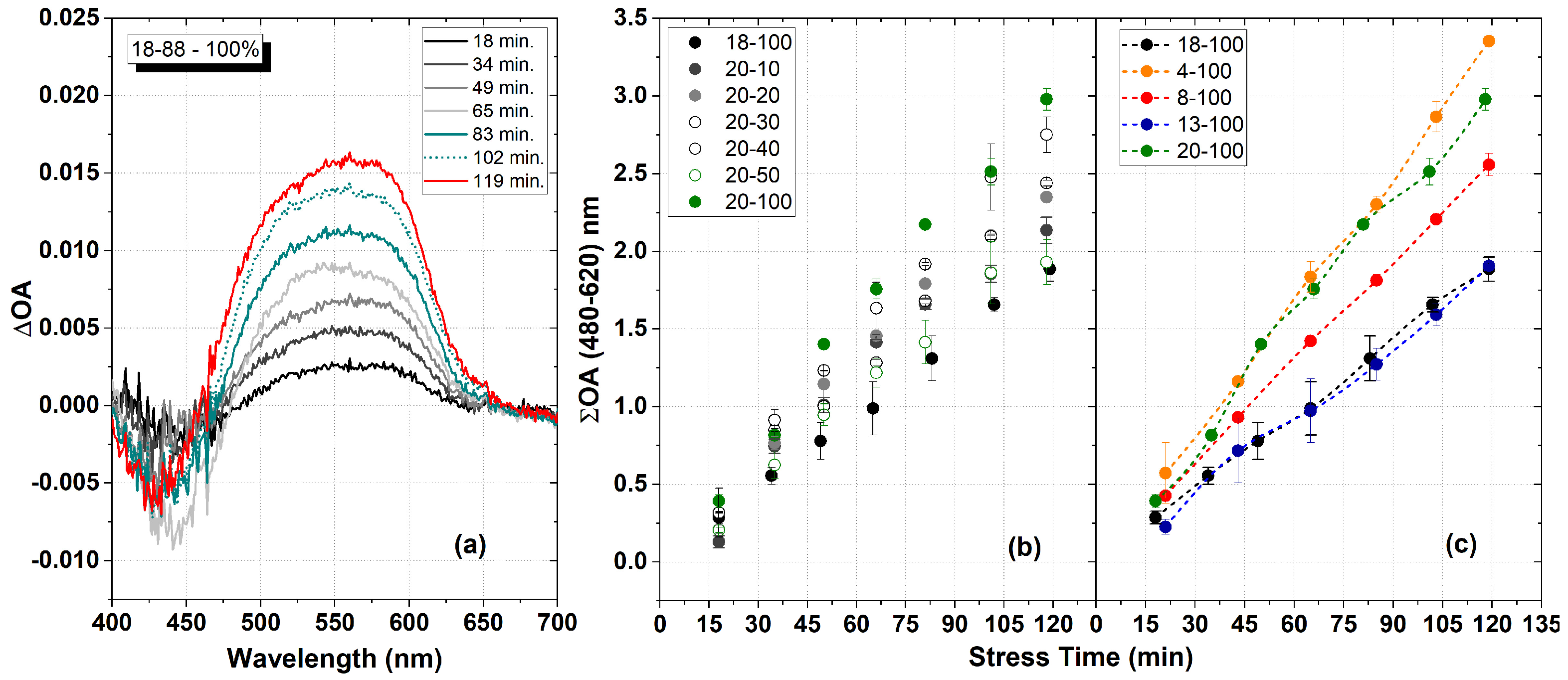
2.5. Dosimetric Analysis
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Hydrogels Preparation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | PVA Mixture | ||||
|---|---|---|---|---|---|
| % 4:88 MW~31 kDa HD~88% | % 8:88 MW~67 kDa HD~88% | % 13:88 MW~94 kDa HD~88% | % 20:98 MW~125 kDa HD~98% | % 18:88 MW~130 kDa HD~88% | |
| 4-100 | 100 | ||||
| 8-100 | 100 | ||||
| 13-100 | 100 | ||||
| 18-100 | 100 | ||||
| 20-100 | 100 | ||||
| 4-10 | 10 | 90 | |||
| 4-20 | 20 | 80 | |||
| 4-30 | 30 | 70 | |||
| 4-40 | 40 | 60 | |||
| 4-50 | 50 | 50 | |||
| 8-10 | 10 | 90 | |||
| 8-20 | 20 | 80 | |||
| 8-30 | 30 | 70 | |||
| 8-40 | 40 | 60 | |||
| 8-50 | 50 | 50 | |||
| 13-10 | 10 | 90 | |||
| 13-20 | 20 | 80 | |||
| 13-30 | 30 | 70 | |||
| 13-40 | 40 | 60 | |||
| 13-50 | 50 | 50 | |||
| 20-10 | 10 | 90 | |||
| 20-20 | 20 | 80 | |||
| 20-30 | 30 | 70 | |||
| 20-40 | 40 | 60 | |||
| 20-50 | 50 | 50 | |||
4.3. Determination of the Viscosity
4.4. Uniaxial Compression Mechanical Tests
4.5. FTIR Measurements
4.6. Gel Fraction Determination
4.7. Swelling Degree Measurements
4.8. Water Loss Determination
4.9. Contact Angle Analyses
4.10. 1H NMR Relaxometry Measurements
4.11. Dosimetric Application
4.11.1. Fricke Hydrogel Dosimeter Preparation
4.11.2. Fricke Hydrogel Dosimeter Irradiation
4.11.3. Optical Absorbance Measurements
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cao, H.; Duan, L.; Zhang, Y.; Cao, J.; Zhang, K. Current Hydrogel Advances in Physicochemical and Biological Response-Driven Biomedical Application Diversity. Signal Transduct. Target. Ther. 2021, 6, 426. [Google Scholar] [CrossRef]
- Correa, S.; Grosskopf, A.K.; Lopez Hernandez, H.; Chan, D.; Yu, A.C.; Stapleton, L.M.; Appel, E.A. Translational Applications of Hydrogels. Chem. Rev. 2021, 121, 11385–11457. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Xia, Z.; Cai, K. Recent Advances in 3D Hydrogel Culture Systems for Mesenchymal Stem Cell-Based Therapy and Cell Behavior Regulation. J. Mater. Chem. B 2022, 10, 1486–1507. [Google Scholar] [CrossRef] [PubMed]
- Nathan, K.G.; Genasan, K.; Kamarul, T. Polyvinyl Alcohol-Chitosan Scaffold for Tissue Engineering and Regenerative Medicine Application: A Review. Mar. Drugs 2023, 21, 304. [Google Scholar] [CrossRef] [PubMed]
- Zulkiflee, I.; Fauzi, M.B. Gelatin-Polyvinyl Alcohol Film for Tissue Engineering: A Concise Review. Biomedicines 2021, 9, 979. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Xiao, R.; Wu, Y.; Xu, L. Advances in Tissue Engineering of Gellan Gum-Based Hydrogels. Carbohydr. Polym. 2024, 324, 121484. [Google Scholar] [CrossRef]
- Liu, X.; Liu, J.; Lin, S.; Zhao, X. Hydrogel Machines. Mater. Today 2020, 36, 102–124. [Google Scholar] [CrossRef]
- Du, J.; Ma, Q.; Wang, B.; Sun, L.; Liu, L. Hydrogel Fibers for Wearable Sensors and Soft Actuators. iScience 2023, 26, 106796. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.; Maji, B.; Yadav, H. Progress on Green Crosslinking of Polysaccharide Hydrogels for Drug Delivery and Tissue Engineering Applications. Carbohydr. Polym. 2024, 326, 121584. [Google Scholar] [CrossRef]
- Garg, A.; Agrawal, R.; Singh Chauhan, C.; Deshmukh, R. In-Situ Gel: A Smart Carrier for Drug Delivery. Int. J. Pharm. 2024, 652, 123819. [Google Scholar] [CrossRef]
- De Deene, Y. Radiation Dosimetry by Use of Radiosensitive Hydrogels and Polymers: Mechanisms, State-of-the-Art and Perspective from 3D to 4D. Gels 2022, 8, 599. [Google Scholar] [CrossRef]
- Zhang, P.; Jiang, L.; Chen, H.; Hu, L. Recent Advances in Hydrogel-Based Sensors Responding to Ionizing Radiation. Gels 2022, 8, 238. [Google Scholar] [CrossRef]
- Marrale, M.; D’Errico, F. Hydrogels for Three-Dimensional Ionizing-Radiation Dosimetry. Gels 2021, 7, 74. [Google Scholar] [CrossRef]
- Macchione, M.A.; Lechón Páez, S.; Strumia, M.C.; Valente, M.; Mattea, F. Chemical Overview of Gel Dosimetry Systems: A Comprehensive Review. Gels 2022, 8, 663. [Google Scholar] [CrossRef]
- Chu, K.C.; Jordan, K.J.; Battista, J.J.; Van Dyk, J.; Rutt, B.K. Polyvinyl Alcohol-Fricke Hydrogel and Cryogel: Two New Gel Dosimetry Systems with Low Fe 3+ Diffusion. Phys. Med. Biol. 2000, 45, 955–969. [Google Scholar] [CrossRef]
- Hill, B.; Bäck, S.Å.J.; Lepage, M.; Simpson, J.; Healy, B.; Baldock, C. Investigation and Analysis of Ferrous Sulfate Polyvinyl Alcohol (PVA) Gel Dosimeter. Phys. Med. Biol. 2002, 47, 4233–4246. [Google Scholar] [CrossRef]
- Smith, S.T.; Masters, K.-S.; Hosokawa, K.; Blinco, J.P.; Crowe, S.B.; Kairn, T.; Trapp, J.V. Technical Note: Preliminary Investigations into the Use of a Functionalised Polymer to Reduce Diffusion in Fricke Gel Dosimeters. Med. Phys. 2015, 42, 6798–6803. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Bai, J.; Shao, K.; Tang, W.; Zhao, X.; Lin, D.; Huang, S.; Chen, C.; Ding, Z.; Ye, J. Poly(Vinyl Alcohol) Hydrogels: The Old and New Functional Materials. Int. J. Polym. Sci. 2021, 2021, 2225426. [Google Scholar] [CrossRef]
- Yeom, C.-K.; Lee, K.-H. Pervaporation Separation of Water-Acetic Acid Mixtures through Poly(Vinyl Alcohol) Membranes Crosslinked with Glutaraldehyde. J. Memb. Sci. 1996, 109, 257–265. [Google Scholar] [CrossRef]
- Marini, A.; Lazzeri, L.; Cascone, M.G.; Ciolini, R.; Tana, L.; d’Errico, F. Fricke Gel Dosimeters with Low-Diffusion and High-Sensitivity Based on a Chemically Cross-Linked PVA Matrix. Radiat. Meas. 2017, 106, 618–621. [Google Scholar] [CrossRef]
- Marrale, M.; Collura, G.; Gallo, S.; Nici, S.; Tranchina, L.; Abbate, B.F.; Marineo, S.; Caracappa, S.; D’Errico, F. Analysis of Spatial Diffusion of Ferric Ions in PVA-GTA Gel Dosimeters through Magnetic Resonance Imaging. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2017, 396, 50–55. [Google Scholar] [CrossRef]
- Lazzeri, L.; Marini, A.; Cascone, M.G.; D’Errico, F. Dosimetric and Chemical Characteristics of Fricke Gels Based on PVA Matrices Cross-Linked with Glutaraldehyde. Phys. Med. Biol. 2019, 64, 085015. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.; Gambarini, G.; Veronese, I.; Argentiere, S.; Gargano, M.; Ianni, L.; Lenardi, C.; Ludwig, N.; Pignoli, E.; d’Errico, F. Does the Gelation Temperature or the Sulfuric Acid Concentration Influence the Dosimetric Properties of Radiochromic PVA-GTA Xylenol Orange Fricke Gels? Radiat. Phys. Chem. 2019, 160, 35–40. [Google Scholar] [CrossRef]
- Gallo, S.; Artuso, E.; Brambilla, M.G.; Gambarini, G.; Lenardi, C.; Monti, A.F.; Torresin, A.; Pignoli, E.; Veronese, I. Characterization of Radiochromic Poly(Vinyl-Alcohol)-Glutaraldehyde Fricke Gels for Dosimetry in External x-Ray Radiation Therapy. J. Phys. D Appl. Phys. 2019, 52, 225601. [Google Scholar] [CrossRef]
- Rabaeh, K.A.; Eyadeh, M.M.; Hailat, T.F.; Madas, B.G.; Aldweri, F.M.; Almomani, A.M.; Awad, S.I. Improvement on the Performance of Chemically Cross-Linked Fricke Methylthymol-Blue Radiochromic Gel Dosimeter by Addition of Dimethyl Sulfoxide. Radiat. Meas. 2021, 141, 106540. [Google Scholar] [CrossRef]
- Scotti, M.; Arosio, P.; Brambilla, E.; Gallo, S.; Lenardi, C.; Locarno, S.; Orsini, F.; Pignoli, E.; Pedicone, L.; Veronese, I. How Xylenol Orange and Ferrous Ammonium Sulphate Influence the Dosimetric Properties of PVA–GTA Fricke Gel Dosimeters: A Spectrophotometric Study. Gels 2022, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Sheykholeslami, N.; Parwaie, W.; Vaezzadeh, V.; Babaie, M.; Farzin, M.; Geraily, G.; Karimi, A.H. Dual Application of Polyvinyl Alcohol Glutaraldehyde Methylthymol Blue Fricke Hydrogel in Clinical Practice: Surface Dosimeter and Bolus. Appl. Radiat. Isot. 2023, 197, 110827. [Google Scholar] [CrossRef]
- Welti, S.E.; Taño, J.E.; Gonzales, C.A.B.; Yasuda, H. Investigation on the Long-Term Storage Condition of the PVA-GTA-I Gel Dosimeter. Radiat. Meas. 2022, 159, 106879. [Google Scholar] [CrossRef]
- Almeida, W.d.S.d.; Alves, A.V.S.; Oliveira, W.F.; da Silveira, M.A.L.; de Souza, S.O.; D’Errico, F.; Sussuchi, E.M. Radiochromic Fricke Gels with Eriochrome Cyanine R for Radiotherapy Dosimetry. Radiat. Phys. Chem. 2022, 191, 109830. [Google Scholar] [CrossRef]
- Taño, J.E.; Gonzales, C.A.B.; Saito, A.; Wada, T.; Nagata, Y.; Yasuda, H. Annealing Properties of the PVA-GTA-I Gel Dosimeter. Radiat. Meas. 2021, 149, 106674. [Google Scholar] [CrossRef]
- Rabaeh, K.A.; Hailat, T.F.; Eyadeh, M.M.; Al- Shorman, M.Y.; Aldweri, F.M.; Alheet, S.M.; Madas, B.G.; Awad, S.I. Dosimetric Properties of Sulfosalicylic Acid-Ferrous-Polyvinyl Alcohol-Glutaraldehyde Hydrogel Dosimeters Using Magnetic and Optical Techniques. Radiat. Phys. Chem. 2020, 177, 109106. [Google Scholar] [CrossRef]
- Wang, T.; Turhan, M.; Gunasekaran, S. Selected Properties of PH-sensitive, Biodegradable Chitosan–Poly(Vinyl Alcohol) Hydrogel. Polym. Int. 2004, 53, 911–918. [Google Scholar] [CrossRef]
- Tanpichai, S.; Oksman, K. Cross-Linked Nanocomposite Hydrogels Based on Cellulose Nanocrystals and PVA: Mechanical Properties and Creep Recovery. Compos. Part A Appl. Sci. Manuf. 2016, 88, 226–233. [Google Scholar] [CrossRef]
- Xiang, D.; Cui, Y.; Wan, Z.; Wang, S.; Peng, L.; Liao, Z.; Chen, C.; Liu, W. Study on Swelling, Compression Property and Degradation Stability of PVA Composite Hydrogels for Artificial Nucleus Pulposus. J. Mech. Behav. Biomed. Mater. 2022, 136, 105496. [Google Scholar] [CrossRef] [PubMed]
- Odian, G. Principles of Polymerization; Wiley: Hoboken, NJ, USA, 2004; ISBN 9780471274001. [Google Scholar]
- Takeichi, T.; Furukawa, N. Epoxy Resins and Phenol-Formaldehyde Resins. In Polymer Science: A Comprehensive Reference; Elsevier: Amsterdam, The Netherlands, 2012; pp. 723–751. [Google Scholar]
- Peppas, N. Hydrogels in Pharmaceutical Formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Mansur, H.S.; Sadahira, C.M.; Souza, A.N.; Mansur, A.A.P. FTIR Spectroscopy Characterization of Poly (Vinyl Alcohol) Hydrogel with Different Hydrolysis Degree and Chemically Crosslinked with Glutaraldehyde. Mater. Sci. Eng. C 2008, 28, 539–548. [Google Scholar] [CrossRef]
- Negim, E.; Rakhmetullayeva, R.; Yeligbayeva, G.; Urkimbaeva, P.; Primzharova, S.; Kaldybekov, D.; Khatib, J.; Mun, G.; Craig, W. Improving Biodegradability of Polyvinyl Alcohol/Starch Blend Films for Packaging Applications. Int. J. Basic Appl. Sci. 2014, 3, 263–273. [Google Scholar] [CrossRef]
- Blout, E.R.; Karplus, R. The Infrared Spectrum of Polyvinyl Alcohol. J. Am. Chem. Soc. 1948, 70, 862–864. [Google Scholar] [CrossRef]
- dos Reis, E.F.; Campos, F.S.; Lage, A.P.; Leite, R.C.; Heneine, L.G.; Vasconcelos, W.L.; Lobato, Z.I.P.; Mansur, H.S. Synthesis and Characterization of Poly (Vinyl Alcohol) Hydrogels and Hybrids for RMPB70 Protein Adsorption. Mater. Res. 2006, 9, 185–191. [Google Scholar] [CrossRef]
- Kudo, S.; Otsuka, E.; Suzuki, A. Swelling Behavior of Chemically Crosslinked PVA Gels in Mixed Solvents. J. Polym. Sci. Part B Polym. Phys. 2010, 48, 1978–1986. [Google Scholar] [CrossRef]
- Okay, O. General Properties of Hydrogels. In Hydrogel Sensors and Actuators; Springer: Berlin/Heidelberg, Germany, 2009; pp. 1–14. [Google Scholar]
- Ottenbrite, R.M.; Park, K.; Okano, T. (Eds.) Biomedical Applications of Hydrogels Handbook; Springer: New York, NY, USA, 2010; ISBN 978-1-4419-5918-8. [Google Scholar]
- Hoti, G.; Caldera, F.; Cecone, C.; Rubin Pedrazzo, A.; Anceschi, A.; Appleton, S.L.; Khazaei Monfared, Y.; Trotta, F. Effect of the Cross-Linking Density on the Swelling and Rheological Behavior of Ester-Bridged β-Cyclodextrin Nanosponges. Materials 2021, 14, 478. [Google Scholar] [CrossRef] [PubMed]
- Goudar, N.; Vanjeri, V.N.; Dixit, S.; Hiremani, V.; Sataraddi, S.; Gasti, T.; Vootla, S.K.; Masti, S.P.; Chougale, R.B. Evaluation of Multifunctional Properties of Gallic Acid Crosslinked Poly (Vinyl Alcohol)/Tragacanth Gum Blend Films for Food Packaging Applications. Int. J. Biol. Macromol. 2020, 158, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Fantazzini, P. Magnetic Resonance for Fluids in Porous Media at the University of Bologna. Magn. Reson. Imaging 2005, 23, 125–131. [Google Scholar] [CrossRef]
- Arosio, P.; Comito, G.; Orsini, F.; Lascialfari, A.; Chiarugi, P.; Ménard-Moyon, C.; Nativi, C.; Richichi, B. Conjugation of a GM3 Lactone Mimetic on Carbon Nanotubes Enhances the Related Inhibition of Melanoma-Associated Metastatic Events. Org. Biomol. Chem. 2018, 16, 6086–6095. [Google Scholar] [CrossRef]
- Du, Y.; Wang, R.; Yue, H.; Zhang, Y.; Wu, H.; Wang, W. Dose Response and Stability of Silicone-Based Deformable Radiochromic Dosimeters (FlexyDos3D) Using Spectrophotometer and Flatbed Scanner. Radiat. Phys. Chem. 2020, 168, 108574. [Google Scholar] [CrossRef]
- Alyani Nezhad, Z.; Geraily, G. A Review Study on Application of Gel Dosimeters in Low Energy Radiation Dosimetry. Appl. Radiat. Isot. 2022, 179, 110015. [Google Scholar] [CrossRef] [PubMed]
- Kuraray Europe GmbH. Available online: https://www.kuraray-poval.com/ (accessed on 1 December 2023).
- Locarno, S.; Gallo, S.; Arosio, P.; Biordi, C.; Dallasega, D.; Gargano, M.; Ludwig, N.; Orsini, F.; Pignoli, E.; Veronese, I.; et al. Dosimetric Double Network Hydrogel Based on Poly(Vinyl-Alcohol)/Phenylalanine-Derivatives with Enhanced Mechanical Properties. ACS Appl. Polym. Mater. 2023, 5, 1902–1914. [Google Scholar] [CrossRef]
- Gallo, S.; Locarno, S.; Brambilla, E.; Lenardi, C.; Pignoli, E.; Veronese, I. Dosimetric Characterization of Double Network Fricke Hydrogel Based on PVA-GTA and Phenylalanine Peptide Derivative. J. Phys. D Appl. Phys. 2024, 57, 075303. [Google Scholar] [CrossRef]
- Gallo, S.; Lizio, D.; Monti, A.F.; Veronese, I.; Brambilla, M.G.; Lenardi, C.; Torresin, A.; Gambarini, G. Temperature Behavior of Radiochromic Poly(Vinyl-Alcohol)-Glutaraldehyde Fricke Gel Dosimeters in Practice. J. Phys. D. Appl. Phys. 2020, 53, 365003. [Google Scholar] [CrossRef]
- Davies, J.B.; Baldock, C. Sensitivity and Stability of the Fricke–Gelatin–Xylenol Orange Gel Dosimeter. Radiat. Phys. Chem. 2008, 77, 690–696. [Google Scholar] [CrossRef]


| Young’s Modulus | Sample ID | ||||
|---|---|---|---|---|---|
| 4-100 | 8-100 | 13-100 | 18-100 | 20-100 | |
| Mean ± 1SD [kPa] | 14.1 ± 3.2 | 23.2 ± 3.3 | 39.6 ± 4.0 | 38.3 ± 5.7 | 72.6 ± 4.2 |
| Sample ID | Contact Angle ± SD (°) |
|---|---|
| 18-100 | 57.78 ± 6.36 |
| 4-100 | 44.61 ± 5.78 |
| 4-50 | 37.02 ± 4.58 |
| 8-100 | 59.48 ± 11.77 |
| 8-50 | 55.61 ± 11.90 |
| 13-100 | 54.07 ± 1.65 |
| 13-50 | 68.55 ± 6.68 |
| 20-100 | 49.65 ± 5.86 |
| 20-50 | 64.78 ± 8.91 |
| Sample ID | T1 ± SD (s) |
|---|---|
| 18-100 | 1.535 ± 0.123 |
| 4-100 | 1.563 ± 0.125 |
| 4-50 | 1.588 ± 0.127 |
| 8-100 | 1.550 ± 0.124 |
| 8-50 | 1.567 ± 0.125 |
| 13-100 | 1.460 ± 0.117 |
| 13-50 | 1.411 ± 0.113 |
| 20-100 | 1.411 ± 0.113 |
| 20-50 | 1.468 ± 0.117 |
| Mean | 1.506 ± 0.023 |
| Sample ID | T2 fast ± SD (s) | Weight of T2 fast (%) | T2 medium ± SD (s) | Weight of T2 medium (%) | T2 slow ± SD (s) | Weight of T2 slow (%) |
|---|---|---|---|---|---|---|
| 18-100 | 0.036 ± 0.003 | 5.3 | 0.626 ± 0.050 | 14.6 | 1.667 ± 0.134 | 80.1 |
| 4-100 | 0.021 ± 0.002 | 4.7 | - | - | 1.601 ± 0.128 | 95.3 |
| 4-50 | 0.009 ± 0.001 | 2.9 | - | - | 1.686 ± 0.135 | 97.1 |
| 8-100 | 0.025 ± 0.002 | 5.0 | 0.638 ± 0.051 | 18.7 | 1.726 ± 0.138 | 76.3 |
| 8-50 | 0.018 ± 0.002 | 6.0 | 0.326 ± 0.026 | 3.7 | 1.631 ± 0.130 | 90.3 |
| 13-100 | 0.018 ± 0.002 | 4.9 | 0.445 ± 0.036 | 0.5 | 1.545 ± 0.124 | 94.6 |
| 13-50 | 0.009 ± 0.001 | 3.7 | - | - | 1.565 ± 0.125 | 96.3 |
| 20-100 | 0.019 ± 0.002 | 4.8 | - | - | 1.140 ± 0.091 | 95.2 |
| 20-50 | 0.021 ± 0.002 | 4.6 | - | - | 1.347 ± 0.108 | 95.4 |
| Sample ID | T2 fast (s) | Weight of T2 fast (%) | T2 medium (s) | Weight of T2 medium (%) | T2 slow (s) | Weight of T2 slow (%) |
|---|---|---|---|---|---|---|
| 18-100 | 0.019 | 4.7 | 0.289 | 6.4 | 1.550 | 88.9 |
| 4-100 | 0.017 | 5.1 | - | - | 1.600 | 94.9 |
| 4-50 | Not Resolved | |||||
| 8-100 | 0.021 | 5.1 | 0.525 | 12.7 | 1.630 | 82.2 |
| 8-50 | 0.013 | 7.5 | 0.332 | 3.4 | 1.630 | 89.1 |
| 13-100 | 0.019 | 4.4 | - | - | 1.540 | 95.6 |
| 13-50 | 0.009 | 3.3 | - | - | 1.560 | 96.7 |
| 20-100 | 0.017 | 4.8 | - | - | 1.140 | 95.2 |
| 20-50 | 0.014 | 4.9 | - | - | 1.320 | 95.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Locarno, S.; Arosio, P.; Curtoni, F.; Piazzoni, M.; Pignoli, E.; Gallo, S. Microscopic and Macroscopic Characterization of Hydrogels Based on Poly(vinyl-alcohol)–Glutaraldehyde Mixtures for Fricke Gel Dosimetry. Gels 2024, 10, 172. https://doi.org/10.3390/gels10030172
Locarno S, Arosio P, Curtoni F, Piazzoni M, Pignoli E, Gallo S. Microscopic and Macroscopic Characterization of Hydrogels Based on Poly(vinyl-alcohol)–Glutaraldehyde Mixtures for Fricke Gel Dosimetry. Gels. 2024; 10(3):172. https://doi.org/10.3390/gels10030172
Chicago/Turabian StyleLocarno, Silvia, Paolo Arosio, Francesca Curtoni, Marco Piazzoni, Emanuele Pignoli, and Salvatore Gallo. 2024. "Microscopic and Macroscopic Characterization of Hydrogels Based on Poly(vinyl-alcohol)–Glutaraldehyde Mixtures for Fricke Gel Dosimetry" Gels 10, no. 3: 172. https://doi.org/10.3390/gels10030172
APA StyleLocarno, S., Arosio, P., Curtoni, F., Piazzoni, M., Pignoli, E., & Gallo, S. (2024). Microscopic and Macroscopic Characterization of Hydrogels Based on Poly(vinyl-alcohol)–Glutaraldehyde Mixtures for Fricke Gel Dosimetry. Gels, 10(3), 172. https://doi.org/10.3390/gels10030172