Abstract
A self-healing gel with self-healing kinetics that can be regulated by heat is developed. The gel is composed of a polymer having benzophenone (BP) substituents, which are cross-linked with a main alkyl chain via ester bonds, titanium chloride, and zinc. This gel material shows a self-healing property at room temperature. Also, its self-healing behavior can be accelerated by heating the gel. This gel having self-healing kinetics that can be regulated by heat is favorable for practical use. When we want to use a self-healing property as a stop-gap measure, a rapid self-healing property is demanded. On the other hand, when we want materials repaired beautifully or decomposed surfaces need to be attached beautifully, a slow self-healing property is favorable. These opposite demands can be answered by the gel with self-healing kinetics that can be regulated by heat.
1. Introduction
Gel materials are composed of polymeric networks and solvents (water [1,2,3], organic solvents [4,5,6], or ionic liquids [7,8,9]) and have high elasticity and softness [10,11,12,13]. Because of these characteristics, gel materials are expected materials to be used for constructing soft robots [14,15], artificial muscles [16,17], adhesives [18,19,20,21], and others [22,23,24,25,26,27].
Among the gel materials, stimuli-responsive gel materials [28,29] are expected to be used for artificial actuators, stimuli-responsive smoke screens, and so on. Due to their usefulness, various stimuli-responsive materials have been enthusiastically studied and reported. For example, photo-responsive gel materials can be formed by using polymers having the side chains interacting via host–guest interactions [30]. The gel composed of a polymer having cyclodextrins and a polymer having azobenzene moieties as side chains shows photo-responsive sol-gel transitions, adhesion, and actuating motion.
Recently, gel materials having self-healing abilities were developed for creating materials having a long lifespan [31,32,33,34]. For example, self-healing gel materials using host–guest interaction between cyclodextrins and their guest compounds were reported [35].
It is possible to reduce consuming ingredients by using self-healing materials having long lifespan, and these materials will help us create an ecofriendly world.
When we design and develop self-healing materials, the kinetics control of self-healing by stimuli, such as photo and heat, is important. Materials for which self-healing kinetics can be controlled by stimuli will help us use their self-healing properties more conveniently. It is favorable to self-heal rapidly when the decomposed materials need to be healed rapidly. On the other hand, it is favorable to self-heal slowly when the decomposed materials need to be healed accurately.
In this research, a self-healing material for which self-healing kinetics can be controlled by heat using a polymer that has ester substituents as side chains and titanium chloride (TiCl4) and zinc was developed and reported. These self-healing materials help us use them by adjusting their self-healing kinetics to the objectives for which they are used.
2. Results and Discussion
2.1. Development and Fabrication of Self-Healing Gels
First, a BP polymer, which has benzophenone substituents as side chains, was synthesized via the free radical polymerization of BP, for which synthetic detail is shown in the experimental section, and acrylamide with 2,2′-(diazene-1,2-diyl)bis(2-ethylpropaneitrile) (AIBN) as a radical initiator in dimethylsulfoxide (DMSO) as shown in Scheme 1 and as described in the Experimental section.
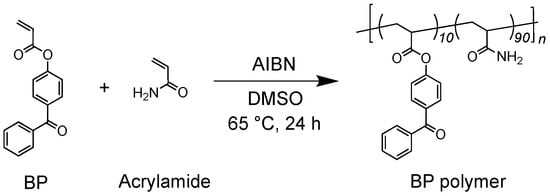
Scheme 1.
Synthesis of BP polymer composed of BP and acrylamide. The synthesized BP polymer was a random copolymer composed of 10 mol% of BP and 90 mol% of acrylamide.
The synthesized BP polymer (0.3 g) was mixed with titanium chloride (TiCl4, 0.5 M) and zinc powder (1.0 M) in 1.0 mL of DMSO. Afterwards, it was confirmed that the mixture gelated as shown in Figure 1. In the mixture, TiCl4 and H2O reacted and released a proton (H+) as shown in Figure 1a. Simultaneously, zinc reacted with TiCl4 and formed a zinc cation (Zn2+) as shown in Figure 1b.
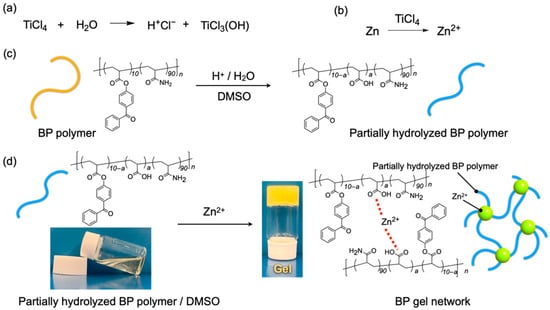
Figure 1.
(a) Reaction of TiCl4 with H2O existing in the air and in dimethylsulfoxide (DMSO), and (b) formation of zinc ion (Zn2+) by the reaction of zinc and TiCl4. (c) Scheme of forming partially hydrolyzed BP polymer from BP polymer by the hydrolysis reaction with HCl formed in (a). (d) Photograph of formation of BP gel network by coordination bonds between partially hydrolyzed BP polymer and zinc ion (Zn2+) formed in (b). The gel was composed of 19 wt% of BP polymer, 0.5 M of TiCl4, and 1.0 M of zinc.
The ester linkages of the BP polymer were hydrolyzed partially by the protons formed in Figure 1a as shown in Figure 1c. The resultant partially hydrolyzed BP polymer having carboxylic acids was cross-linked by Zn2+ via a coordinate bond [36,37] as shown in Figure 1d. It was considered that the gelation was carried out by these reactions of the BP polymer, TiCl4, and zinc.
2.2. SEM Observation of Self-Healing Gels
Scanning electron microscope (SEM) images were observed to evaluate the morphology of the gel as shown in Figure 2. To remove DMSO, the prepared gel was soaked into ion-exchanged water for 24 h twice to exchange the solvent from DMSO to water. After that, the gel was lyophilized as shown in Figure 2a and used for SEM observation. The obtained SEM images showed that the BP polymer formed a network structure as shown in Figure 2b,c. It is considered that the BP polymers were cross-linked by metal ions and formed a stable gel network.

Figure 2.
(a) Photograph of the xerogel prepared using the gel composed of BP polymer, TiCl4, and zinc. The original BP gel before exchanging the solvent from DMSO to water was composed of 19 wt% of BP polymer, 0.5 M of TiCl4 and 1.0 M of zinc. (b,c) Scanning electron microscope images of BP xerogel. The white rectangle dashed line in (b) shows the observation area of (c).
2.3. Self-Healing Ability of Gels
The gel composed of 19 wt% of BP polymer, 0.5 M of TiCl4, and 1.0 M of zinc was cut into two pieces with a razor as shown in Figure 3. After that, the cut pieces were brought into contact with each other and were left to stand for 24 h at room temperature. Consequently, the cut pieces self-healed and formed an original gel again as shown in Figure 3a. The self-healed gel could be pulled with pairs of tweezers without separating again.
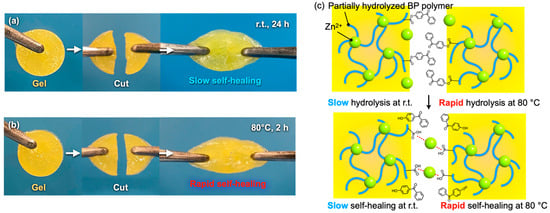
Figure 3.
Photographs of the self-healing test of the gels composed of 19 wt% of BP polymer, 0.5 M of TiCl4, 1.0 M of zinc, and 1.0 mL of DMSO at (a) room temperature and at (b) 80 °C. (c) Schematic illustration of self-healing mechanism and difference of self-healing kinetics between room temperature and 80 °C.
The gel pieces were heated at 80 °C in the same manner as at room temperature. The gel pieces were self-healed and formed one gel piece again more rapidly at 80 °C than at room temperature as shown in Figure 3b. It was confirmed that the cut gel pieces adhered again in 2 h. Considering that the gel self-healed after 24 h at room temperature, the gel self-healed much more rapidly at 80 °C. In this way, the self-healing could be accelerated by heating the cut gel pieces after they were brought into contact with each other. It is considered that the difference of the self-healing kinetics between room temperature and 80 °C was caused by the difference in the hydrolysis kinetics of the ester groups as shown in Figure 3c. The hydrolysis reaction of the ester groups linking between benzophenone and the polymer chain were more rapid at 80 °C than at room temperature and made the gel networks cross-linked again rapidly.
2.4. Self-Healing Test of Reference Samples
To evaluate the self-healing mechanism of the gel, some reference samples were prepared as shown in Figure 4. Firstly, the polyacrylamide was used for preparing a sample instead of the BP polymer. The prepared sample did not gelate as shown in Figure 4a. It was considered that the acrylamide polymer not having ester groups did not form carboxylic acid groups, which interact with zinc ions even after mixing TiCl4 and zinc. Hence, the sample did not form a gel network. Second, a sample was composed of a polymer synthesized from acrylic acid and acrylamide (AAc polymer), TiCl4, and zinc. The polymer did not contain ester groups in its chemical structure and had a structure in which the BP substituents were removed from the structure of the BP polymer. After mixing all the components, the solution gelated. However, the gel did not have a self-healing ability as shown in Figure 4b. It is supposed that the gel composed of the AAc polymer, TiCl4, and zinc did not self-heal because the AAc polymers had too many carboxylic acid groups, which could interact with zinc ions, and there were few free zinc cations, which could interact with the carboxylic acid of the AAc polymers and be used for self-healing in the gel. From the result, it is considered that the degree of the hydrolysis of ester linkages affects the self-healing ability and mechanical strength critically.
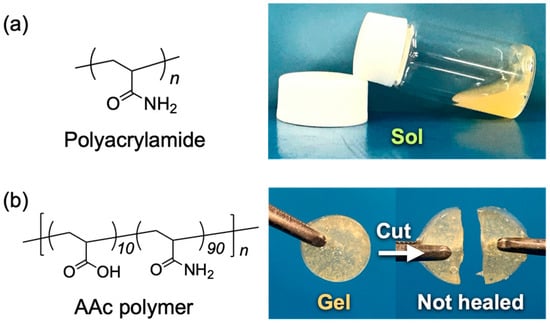
Figure 4.
(a) Chemical structure of polyacrylamide and photograph of a DMSO solution of 19 wt% of polyacrylamide mixed with TiCl4 (0.5 mM) and zinc powder (1.0 M). (b) Chemical structure of AAc polymer and photograph of a DMSO gel formed by mixing 19 wt% of AAc polymer with TiCl4 (0.5 M) and zinc powder (1.0 M). The resultant gel did not have self-healing ability like BP gel, and the cut gel pieces were separated easily by pulling them even after 48 h.
2.5. Indentation Test
An indentation test was carried out to evaluate the mechanical strength of the self-healed interfaces as shown in Figure 5. Figure 5a shows the dependency of the mechanical strength of the self-healed interfaces on the self-healing time at room temperature. As can be seen, the mechanical strength increased as the self-healing time increased. The concentration dependency of the self-healing rates calculated from the obtained results of Figure 5a was shown in Figure 5b. The self-healing rates were calculated from the inclination of displacement-stress curves of the original gels and the self-healed gels. The self-healing rates of the self-healed interfaces increased as the concentrations of the carboxylic acid in the gel increased.
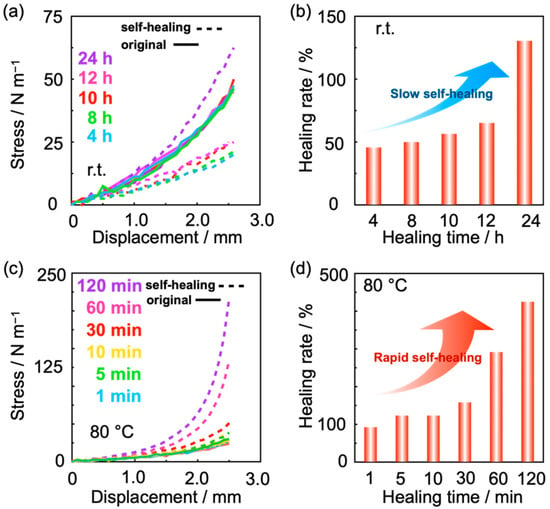
Figure 5.
(a) Displacement and stress curves of the DMSO gel composed of BP polymer, TiCl4, and zinc before and after self-healing at room temperature. The rigid lines show the results of the original BP gels, and the dash lines show those of the self-healed gels. (b) Self-healing rate depending on the self-healing time at room temperature calculated from (a). Self-healing rate was the value that the inclination of the curve of self-healed interface divided with that of the original gel. (c) Displacement and stress curves of the DMSO gel composed of BP polymer, TiCl4, and zinc before and after self-healing at 80 °C. (d) Self-healing rate depending on the self-healing time at 80 °C calculated from (c). The bold lines and the dash lines in (a,c) showed the results of the original gels, and those of the self-healed gels, respectively. The gels used for the experiments were composed of 19 wt% of BP polymer, 0.5 M of TiCl4, and 1.0 M of zinc.
Time-course changes of the mechanical strength of the self-healing interfaces were measured at 80 °C. As can be seen in Figure 5c,d, the mechanical strength of the self-healing interfaces recovered rapidly at 80 °C compared with room temperature. In this way, the results of the indentation test indicated that the interfaces self-healed rapidly by heating. After 5 min of self-healing time, the self-healing efficiency was over 100% because the hydrolysis of ester groups progressed, and the resultant carboxylic acids interacted with zinc ions more than before the self-healing experiment. It is supposed that the difference in the self-healing kinetics is due to the difference of the hydrolysis reaction progress at room temperature and 80 °C.
2.6. 1H NMR Spectroscopy
1H NMR spectroscopy was carried out to evaluate the mechanism of the self-healing phenomenon of the gel composed of the BP polymer, TiCl4, and zinc as shown in Figure 6 and Figure 7. Figure 6 showed the result of 1H NMR spectroscopy at room temperature. The pattern of the signals assigned to the protons of the aromatic groups of the BP polymer around 6.6–7.8 ppm changed as time passed because the ester groups were decomposed by TiCl4. In particular, the signal intensity at 6.9 ppm increased because 4-hydroxyacetophenone formed by the hydrolysis with TiCl4. The formed multiple carboxylic acid interacted with zinc cations, and then the polymers were cross-linked via the coordination bonds between formed carboxylic acids and zinc cations. Figure 7 showed the result of 1H NMR spectroscopy at 80 °C. As can be seen in the figure, the hydrolysis reaction was accelerated by heating at 80 °C. The signals assigned to 4-hydroxybenzophenone were increased much faster and were seen more sharply and clearly than at room temperature. The difference in changes of the NMR spectra based on temperature indicates that the self-healing kinetics can be regulated by heat.
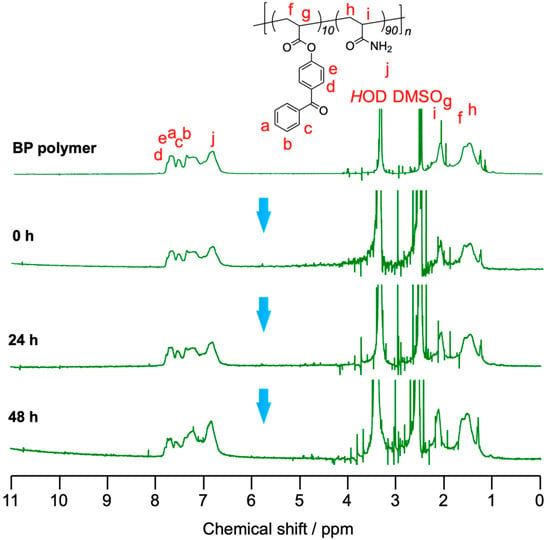
Figure 6.
Time-course change of 1H NMR spectra (500 MHz, DMSO-d6, r.t.) of BP polymer (19 wt%) mixed with TiCl4 (0.5 M) and zinc powder (1.0 M) in DMSO at room temperature.
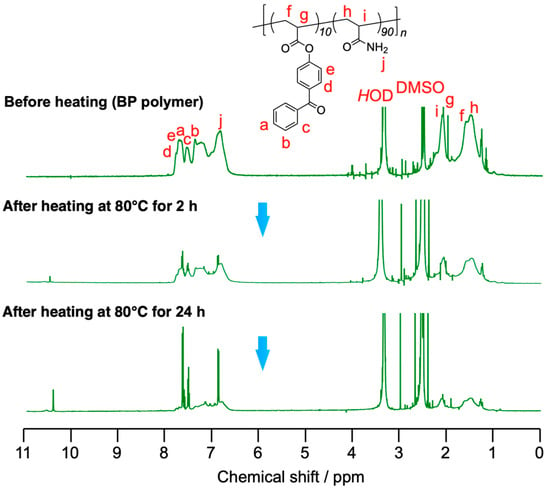
Figure 7.
Time-course change of 1H NMR spectra (500 MHz, DMSO-d6, r.t.) of 19 wt% of BP polymer mixed with TiCl4 (0.5 M) and zinc powder (1.0 M) in DMSO and heated at 80 °C.
Next, the hydrolysis of the BP polymer was evaluated by using a model compound, 4-acetoxybenzophenone, as shown in Figure S5. Proton signals of 4-hydroxybenzophenone appeared in the 1H NMR spectra of 4-acetoxybenzophenone in deuterated DMSO by 48 h after the BP polymer with TiCl4 and zinc at room temperature and at 80 °C. Stronger proton signals of hydrolyzed 4-hydroxybenzophenone were observed from the sample heated at 80 °C, and it was confirmed that the kinetics of the hydrolysis differed depending on the temperature. These changes also support the hypothesis that the BP polymer formed a gel and showed the self-healing ability due to the hydrolysis of ester linkages.
2.7. Fourier-Transform Attenuated Total Reflection Infrared (FT-ATR-IR) Spectroscopy
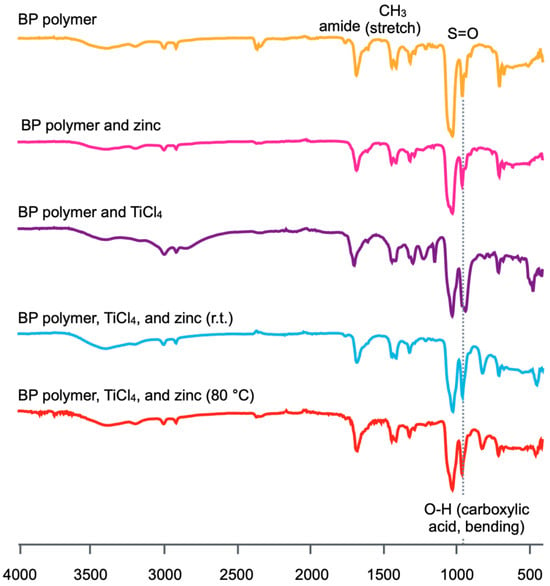
Fourier-transform attenuated total reflection infrared (FT-ATR-IR) spectroscopy was carried out to evaluate the mechanism of the gelation and the self-healing as shown in Figure 8. As can be seen in the figure, there is no significant spectral change in the spectrum of the sample composed of the BP polymer and zinc compared with that of the BP polymer. On the other hand, it was confirmed that the absorption band at 900 cm−1 assigned to the out-on-plane bending motion of the carboxylic acid groups increased in the spectra of the samples mixed with TiCl4. This spectral change means that the carboxylic groups in the BP polymer decomposed to carboxylic acid by the reaction with TiCl4. In this way, the results obtained from IR spectroscopy support the hypothesis that the gel formation and self-healing property of the gels composed of BP polymer, TiCl4, and zinc were due to the hydrolysis of ester groups by TiCl4.
3. Conclusions
Controlling self-healing kinetics is important when self-healing materials are intended for practical use. Slow self-healing is favorable when we make materials self-heal accurately so as not to leave any scars on the surfaces. On the other hand, fast self-healing is favorable when we make materials self-heal quickly as a stopgap measure. In this research, we developed a self-healing material with self-healing kinetics that can be regulated by temperature. In this self-healing system, ester bonds cross-linking between a polymer main chain and benzophenone substituents were hydrolyzed depending on temperature and formed carboxyl acid groups at the main chains. These carboxy acid groups interacted with zinc ions and bridged polymer chains via coordinal bonds. It is expected that the self-healing materials with controllable kinetics reported in this manuscript would help us construct a sustainable and eco-friendly society using self-healing materials having long lifespan because the self-healing kinetics can be changed depending on objectives for which they are used. Moreover, it is expected that it will become possible to make artificial skins having more superior self-healing abilities than the skin of our human bodies that cannot control the kinetics of self-healing. This can be done by continuing the studies of the self-healing materials for which self-healing kinetics are controllable like the gel reported in this research.
4. Materials and Methods
4.1. Materials and Measurement Apparatus
Acrylamide (AAm), triethylamine, acetone, ethylacetate, tetrahydrofuran (THF), 2,2′-(diazene-1,2-diyl)bis(2-ethylpropaneitrile) (AIBN), hydrochloric acid (HCl), sodium chloride (NaCl), magnesium sulfate (anhydrous), dimethylsulfoxide (DMSO), DMSO-d6, and chloroform-d1 were purchased from Kanto Chemical Co. Inc., Tokyo, Japan. The 4-hydroxybenzophenone, acryloyl chloride, and zinc (powder) were purchased from Tokyo Chemical Industry (TCI), Tokyo, Japan. All other reagents were purchased from Kanto Chemical Co. Inc., Tokyo, Japan. 1H NMR and 13C NMR spectra were recorded on a Varian model 500-MR spectrometer, Los Angeles, CA, USA. The indentation test was performed using Imada MX2-500N-FA, Toyohashi, Aichi, Japan, equipped with Imada ZTA-50N, Toyohashi, Aichi, Japan. The rate of extending samples was 10 mm/min. Infrared (IR) spectroscopy was conducted using a JASCO FT/IR-4100, Tokyo, Japan. Scanning electron microscopy (SEM) images were recorded using JEOL JCM-5610LV scanning electron microscope, Tokyo, Japan. SEM samples were spin-coated with platinum using an ion sputter coater (Hitachi E-1030 Ion Sputter Coater), Tokyo, Japan.
4.2. Synthesis of 4-Acryloylbenzophenone (BP)
4-acryloylbenzophenone (BP) was synthesized according to the literature previously reported [38,39]. The 4-hydroxybenzophenone (25 mmol) and triethylamine (36 mmol) were dissolved into 25 mL of dried tetrahydrofuran (THF) in a 100 mL round bottom flask. The solution was cooled in an ice bath. Then, 25 mL of dried THF solution, in which acryloylchloride (50 mmol) was dissolved, was added to the solution dropwise, and the solution was stirred for 30 min. After that, the solution was filtrated, and the filtrate was collected. The solution was mixed with ethylacetate and water and extracted. The collected organic layer was washed with 0.1 M hydrogen chloride (HCl) aqueous solution and brine three times each. After magnesium sulfoxide was added to the solution, the solution was filtrated, and its solvent was evaporated. The compound was estimated as BP by 1H NMR spectroscopy as shown in Figure S1. The yield of the reaction was 84%.
4.3. Synthesis of BP Polymer
BP (10 mmol), AAm (90 mmol), and AIBN (12 mmol) were dissolved into 75 mL of DMSO in a 200 mL round bottom flask. After N2 gas was purged into the flask, the solution was heated at 65 °C for 10 h. Consequently, a clear viscous solution was obtained. After acetone was added to the solution, white precipitation took place. This precipitation was collected by filtration and washed with acetone several times. The resultant precipitation was dried in vacuo and collected as BP polymer. The compound was estimated by 1H NMR spectroscopy as shown in Figure S2.
4.4. Synthesis of Polyacrylamide
AAm (90 mmol) and AIBN (12 mmol) were dissolved into 75 mL of DMSO in a 200 mL round bottom flask. After N2 gas was purged into the flask, the solution was heated at 65 °C for 10 h. After acetone was added to the solution, white precipitation took place. This precipitation was collected by filtration and washed with acetone several times. The resultant precipitation was dried in vacuo and collected as polyacrylamide (pAAm). The compound was estimated by 1H NMR spectroscopy as shown in Figure S3.
4.5. Synthesis of Poly(acrylic acid-co-acrylamide) (AAc Polymer)
Acrylic acid (AAc, 10 mmol), AAm (90 mmol), and 2,2′-azobisisobutyronitrile (AIBN, 12 mmol) were dissolved into 75 mL of DMSO in a 200 mL round bottom flask. After N2 gas was purged into the flask, the solution was heated at 65 °C for 10 h. Consequently, a clear viscous solution was obtained. After acetone was added to the solution, white precipitation took place. This precipitation was collected by filtration and washed with acetone several times. The resultant precipitation was dried in vacuo and collected as poly(acrylic acid-co-acrylamide) (AAc polymer). The compound was estimated by 1H NMR spectroscopy as shown in Figure S4.
4.6. Synthesis of 4-Acetoxybenzophenone
The 4-acetoxybenzophenone used for evaluating the hydrolysis of the BP polymer was synthesized according to the literature previously reported [40].
4.7. Mixing TiCl4 and Zinc in DMSO
Zinc powder (10 mmol) was dispersed in 10 mL of DMSO and stirred at 0 °C for 10 min under N2 atmosphere. Titanium chloride (TiCl4, 5.0 mmol) was added to the solution dropwise and heated at 70 °C for 2 h. The resultant mixture was used for preparing the gel after being cooled to room temperature.
4.8. Preparation of BP Gel
BP polymer (0.3 g) and the prepared 1.0 mL of the DMSO mixture of TiCl4 and zinc were mixed in a sample tube and heated at 80 °C for 24 h. In the resultant mixture, 1.0 M of zinc and 0.5 M of TiCl4 existed. After heating, the mixture formed a gel (BP gel), for which the solvent was DMSO.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/gels10060410/s1, Figure S1: 1H NMR (500 MHz, CDCl3, r.t.) spectrum of 4-acryloylbenzophenone (BP); Figure S2: 1H NMR (500 MHz, DMSO-d6, r.t.) spectrum of BP polymer (10:90 (mol:mol)); Figure S3: 1H NMR (500 MHz, DMSO-d6, r.t.) spectrum of polyacrylamide (pAAm); Figure S4: 1H NMR (500 MHz, DMSO-d6, r.t.) spectrum of AAc polyner (10:90 (mol:mol)); Figure S5: 1H NMR spectra (500 MHz, DMSO-d6, r.t.) of (a) 4-acetoxybenzophenone before mixing with TiCl4 and zinc, and 4-acetoxybenzophenone left to stand for 48 h at (b) room temperature and (c) 80 °C after mixed with 0.5 M of TiCl4 and 1.0 M of zinc.
Author Contributions
Conceptualization, S.T.; methodology, S.T.; synthesis of polymers, R.S.; material analysis, R.S. and S.T.; spectroscopy, R.S. and S.T.; SEM observation, R.S.; indentation test, R.S.; writing—original draft preparation, S.T.; writing—review and editing, R.S.; visualization, R.S. and S.T.; supervision, S.T.; project administration, S.T.; funding acquisition, S.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions of the research are included in the article. Further inquiries can be directed to the corresponding author.
Acknowledgments
We thank Makoto Roppongi, Utsunomiya University, for his kindly experimental support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang, Q.; Mynar, J.L.; Yoshida, M.; Lee, E.; Lee, M.; Okuro, K.; Kinbara, K.; Aida, T. High-Water-Content Mouldable Hydrogels by Mixing Clay and a Dendritic Molecular Binder. Nature 2010, 463, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Naahidi, S.; Jafari, M.; Logan, M.; Wang, Y.; Yuan, Y.; Bae, H.; Dixon, B.; Chen, P. Biocompatibility of Hydrogel-Based Scaffolds for Tissue Engineering Applications. Biotech. Adv. 2017, 35, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Anthis, A.H.C.; Abundo, M.P.; Neuer, A.L.; Tsolaki, E.; Rosendorf, J.; Rduch, T.; Starsich, F.H.L.; Weisse, B.; Liska, V.; Schlegel, A.A.; et al. Modular Stimuli-Responsive Hydrogel Sealants for Early Gastrointestinal Leak Detection and Containment. Nat. Commun. 2022, 13, 7311. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Iwashita, Y.; Shirakawa, M.; Kawano, S.; Fujita, N.; Shinkai, S. Spontaneous Colorimetric Sensing of the Positional Isomers of Dihydroxynaphthalene in a 1D Organogel Matrix. Angew. Chem. 2006, 118, 1622–1625. [Google Scholar] [CrossRef]
- Sugiyasu, K.; Fujita, N.; Shinkai, S. Visible-Light-Harvesting Organogel Composed of Cholesterol-Based Perylene Derivatives. Angew. Chem. Int. Ed. 2004, 43, 1229–1233. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, P.; Liu, M.; Wang, S.; Jiang, L. Organogel-Based Thin Films for Self-Cleaning on Various Surfaces. Adv. Mater. 2013, 25, 4477–4481. [Google Scholar] [CrossRef] [PubMed]
- Susan, M.A.B.H.; Kaneko, T.; Noda, A.; Watanabe, M. Ion Gels Prepared by in Situ Radical Polymerization of Vinyl Monomers in an Ionic Liquid and Their Characterization as Polymer Electrolytes. J. Am. Chem. Soc. 2005, 127, 4976–4983. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Watanabe, M. Macromolecules in Ionic Liquids: Progress, Challenges, and Opportunities. Macromolecules 2008, 41, 3739–3749. [Google Scholar] [CrossRef]
- Fukushima, T.; Kosaka, A.; Ishimura, Y.; Yamamoto, T.; Takigawa, T.; Ishii, N.; Aida, T. Molecular Ordering of Organic Molten Salts Triggered by Single-Walled Carbon Nanotubes. Science 2003, 300, 2072–2074. [Google Scholar] [CrossRef]
- Sun, J.-Y.; Zhao, X.; Illeperuma, W.R.K.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly Stretchable and Tough Hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takehisa, T. Nanocomposite Hydrogels: A Unique Organic–Inorganic Network Structure with Extraordinary Mechanical, Optical, and Swelling/De-Swelling Properties. Adv. Mater. 2002, 14, 1120. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-Network Hydrogels with Extremely High Mechanical Strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Okumura, Y.; Ito, K. The Polyrotaxane Gel: A Topological Gel by Figure-of-Eight Cross-Links. Adv. Mater. 2001, 13, 485–487. [Google Scholar] [CrossRef]
- Lee, Y.; Song, W.J.; Sun, J.-Y. Hydrogel Soft Robotics. Mater. Today Phys. 2020, 15, 100258. [Google Scholar] [CrossRef]
- Calvert, P. Hydrogels for Soft Machines. Adv. Mater. 2009, 21, 743–756. [Google Scholar] [CrossRef]
- Imaizumi, S.; Kokubo, H.; Watanabe, M. Polymer Actuators Using Ion-Gel Electrolytes Prepared by Self-Assembly of ABA-Triblock Copolymers. Macromolecules 2012, 45, 401–409. [Google Scholar] [CrossRef]
- Kim, Y.S.; Liu, M.; Ishida, Y.; Ebina, Y.; Osada, M.; Sasaki, T.; Hikima, T.; Takata, M.; Aida, T. Thermoresponsive Actuation Enabled by Permittivity Switching in an Electrostatically Anisotropic Hydrogel. Nat. Mater. 2015, 14, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Prevoteau, A.; Elzière, P.; Hourdet, D.; Marcellan, A.; Leibler, L. Nanoparticle Solutions as Adhesives for Gels and Biological Tissues. Nature 2014, 505, 382–385. [Google Scholar] [CrossRef]
- Okada, R.; Tamesue, S. Indirect Adhesion of Hydrogels via the Radical Polymerization Mediated by N,N,N′,N′-Tetramethylethylenediamine and Ammonium Persulfate. ACS Appl. Polym. Mater. 2024, 6, 1268–1275. [Google Scholar] [CrossRef]
- Tsurumaki, F.; Sato, A.; Fukai, K.; Maruoka, S.; Tamesue, S. Development of an Adhesive Gel System for Changing the Structure and Properties of Its Adhesive Joint via Reactions with Amine Molecules after Adhesion. ACS Appl. Mater. Interfaces 2023, 15, 32852–32862. [Google Scholar] [CrossRef]
- Cheng, S.; Wang, H.; Pan, X.; Zhang, C.; Zhang, K.; Chen, Z.; Dong, W.; Xie, A.; Qi, X. Dendritic Hydrogels with Robust Inherent Antibacterial Properties for Promoting Bacteria-Infected Wound Healing. ACS Appl. Mater. Interfaces 2022, 14, 11144–11155. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Kawakami, R.; Namba, R.; Nakajima, T.; Gong, J.P. Mechanoresponsive Self-Growing Hydrogels Inspired by Muscle Training. Science 2019, 363, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R. Self-Oscillating Gels Driven by the Belousov-Zhabotinsky Reaction as Novel Smart Materials. Adv. Mater. 2010, 22, 3463–3483. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Viveros, J.A.; Wrue, M.H.; Anthamatten, M. Shape-Memory Effects in Polymer Networks Containing Reversibly Associating Side-Groups. Adv. Mater. 2007, 19, 2851–2855. [Google Scholar] [CrossRef]
- Kumpfer, J.R.; Rowan, S.J. Thermo-, Photo-, and Chemo-Responsive Shape-Memory Properties from Photo-Cross-Linked Metallo-Supramolecular Polymers. J. Am. Chem. Soc. 2011, 133, 12866–12874. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.T.; Hwang, Y.; Chen, A.C.; Varghese, S.; Sah, R.L. Cartilage-like Mechanical Properties of Poly (Ethylene Glycol)-Diacrylate Hydrogels. Biomaterials 2012, 33, 6682–6690. [Google Scholar] [CrossRef] [PubMed]
- Yuk, H.; Zhang, T.; Parada, G.A.; Liu, X.; Zhao, X. Skin-Inspired Hydrogel–Elastomer Hybrids with Robust Interfaces and Functional Microstructures. Nat. Commun. 2016, 7, 12028. [Google Scholar] [CrossRef] [PubMed]
- Shymborska, Y.; Budkowski, A.; Raczkowska, J.; Donchak, V.; Melnyk, Y.; Vasiichuk, V.; Stetsyshyn, Y. Switching It Up: The Promise of Stimuli-Responsive Polymer Systems in Biomedical Science. Chem. Rec. 2023, e202300217. [Google Scholar] [CrossRef] [PubMed]
- Tamesue, S.; Noguchi, S.; Kimura, Y.; Endo, T. Reversing Redox Responsiveness of Hydrogels Due to Supramolecular Interactions by Utilizing Double-Network Structures. ACS Appl. Mater. Interfaces 2018, 10, 27381–27390. [Google Scholar] [CrossRef]
- Tamesue, S.; Takashima, Y.; Yamaguchi, H.; Shinkai, S.; Harada, A. Photoswitchable Supramolecular Hydrogels Formed by Cyclodextrins and Azobenzene Polymers. Angew. Chem. Int. Ed. 2010, 49, 7461–7464. [Google Scholar] [CrossRef]
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-Healing and Thermoreversible Rubber from Supramolecular Assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.L.; in het Panhuis, M. Self-Healing Hydrogels. Adv. Mater. 2016, 28, 9060–9093. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, Q.; Sun, Y.; Bai, H.; Shi, G. Three-Dimensional Self-Assembly of Graphene Oxide and DNA into Multifunctional Hydrogels. ACS Nano 2010, 4, 7358–7362. [Google Scholar] [CrossRef] [PubMed]
- Tamesue, S.; Ohtani, M.; Yamada, K.; Ishida, Y.; Spruell, J.M.; Lynd, N.A.; Hawker, C.J.; Aida, T. Linear versus Dendritic Molecular Binders for Hydrogel Network Formation with Clay Nanosheets: Studies with ABA Triblock Copolyethers Carrying Guanidinium Ion Pendants. J. Am. Chem. Soc. 2013, 135, 15650–15655. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. Redox-Responsive Self-Healing Materials Formed from Host–Guest Polymers. Nat. Commun. 2011, 2, 511. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Peng, Z.; Zhang, Y.; Zhang, Y. In Situ Preparation of Zinc Salts of Unsaturated Carboxylic Acids to Reinforce NBR. J. Appl. Polym. Sci. 2000, 77, 2740–2748. [Google Scholar] [CrossRef]
- Anan, S.; Kurihara, T.; Yamaguchi, M.; Kikuchi, H.; Kokado, K. Enhanced Orientation of Liquid Crystals Inside Micropores of Metal–Organic Frameworks Having Thermoresponsivity. Chem. Eur. J. 2024, 30, e202303277. [Google Scholar] [CrossRef] [PubMed]
- Temel, G.; Enginol, B.; Aydin, M.; Balta, D.K.; Arsu, N. Photopolymerization and Photophysical Properties of Amine Linked Benzophenone Photoinitiator for Free Radical Polymerization. J. Photochem. Photobiol. A 2011, 219, 26–31. [Google Scholar] [CrossRef]
- Jung, S.-H.; Lee, H.T.; Park, M.J.; Lim, B.; Park, B.C.; Jung, Y.J.; Kong, H.; Hwang, D.-H.; Lee, H.; Park, J.M. Precisely Tunable Humidity Color Indicator Based on Photonic Polymer Films. Macromolecules 2021, 54, 621–628. [Google Scholar] [CrossRef]
- Sack, D.; Wagenknecht, H. The Concept of Photozymes: Short Peptides with Photoredox Catalytic Activity for Nucleophilic Additions to α-Phenyl Styrenes. Eur. J. Org. Chem. 2021, 2021, 6400–6407. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).