

Hydrogels from Protein–Polymer Conjugates: A Pathway to Next-Generation Biomaterials

 , , , ,
, , , , 
Abstract
1. Introduction
2. Synthesis of Protein–Polymer Conjugates
2.1. Controlled Radical Polymerization (CRP)
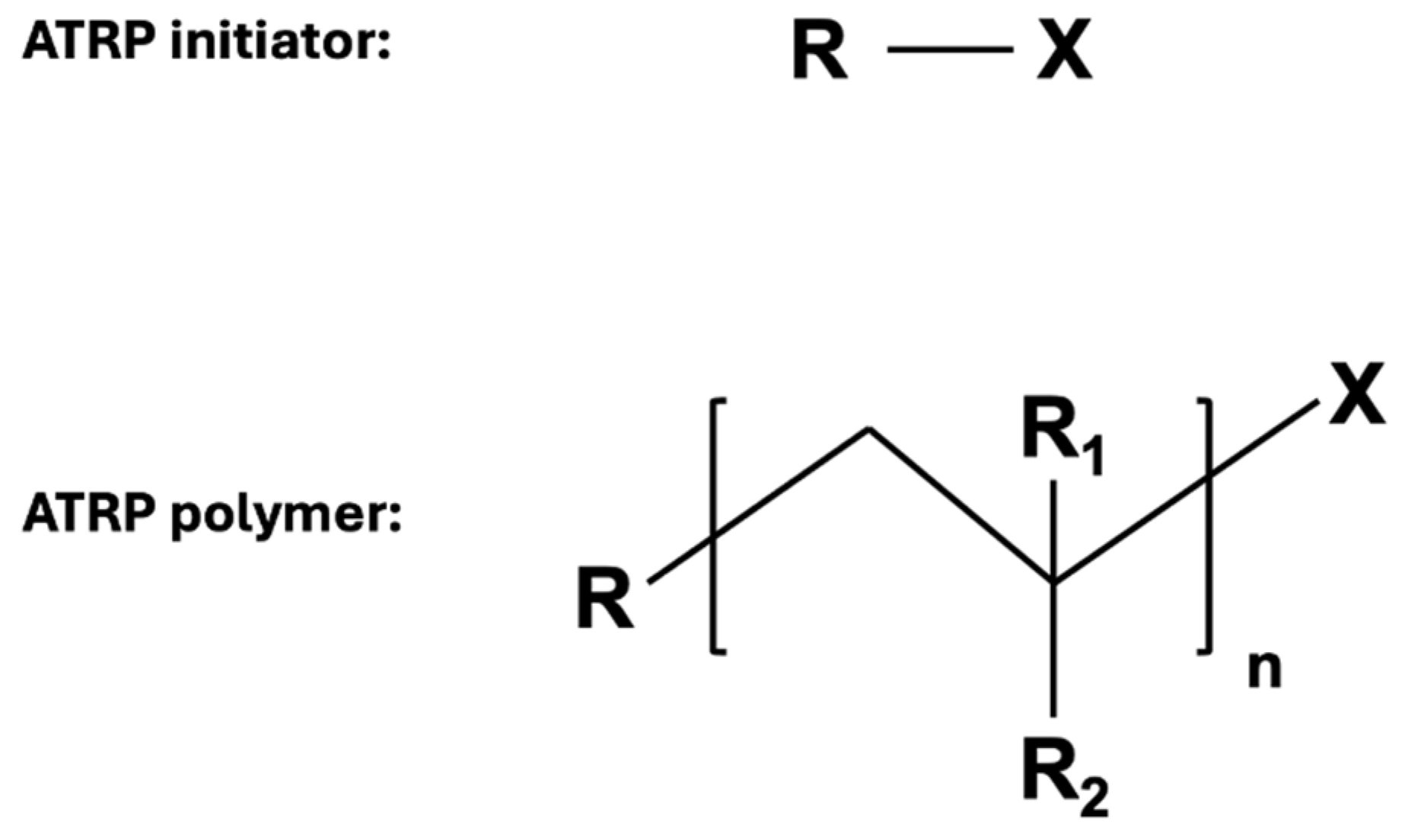
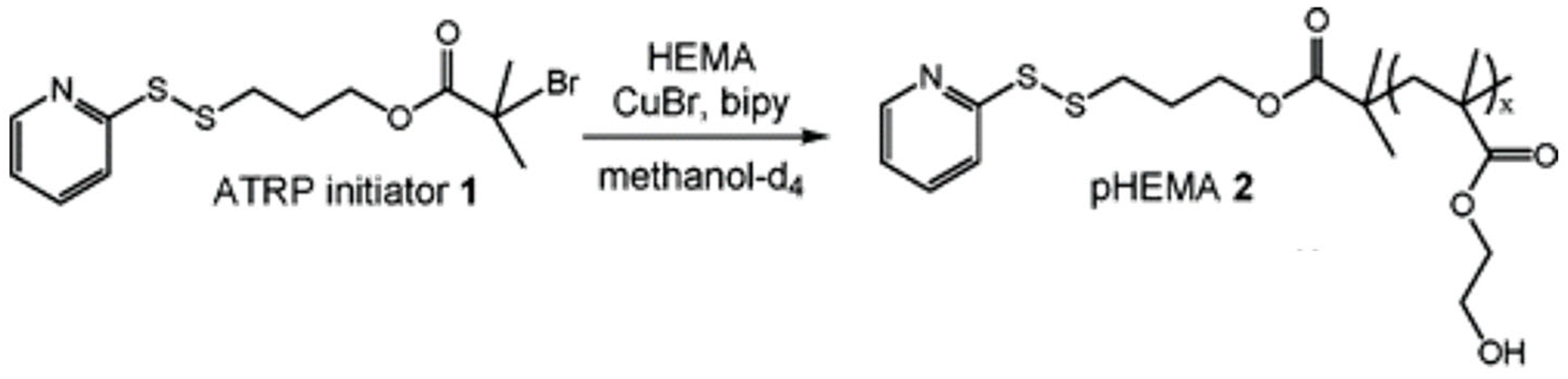
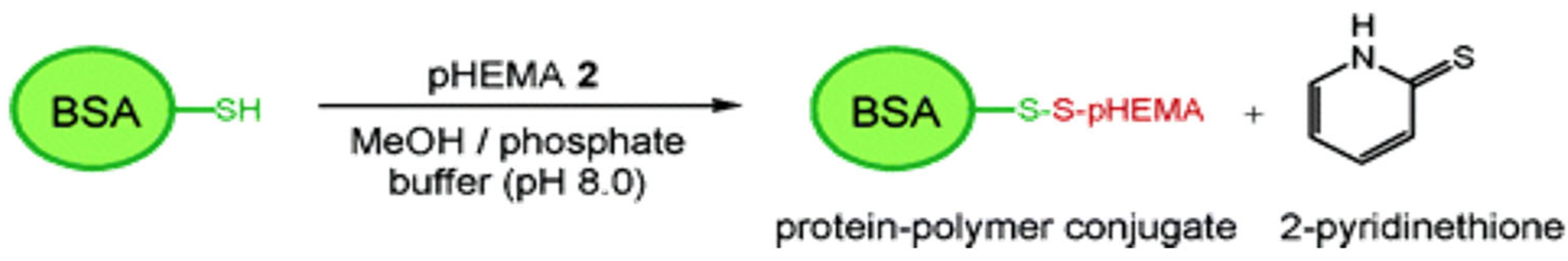
2.1.1. Atom Transfer Radical Polymerization (ATRP)
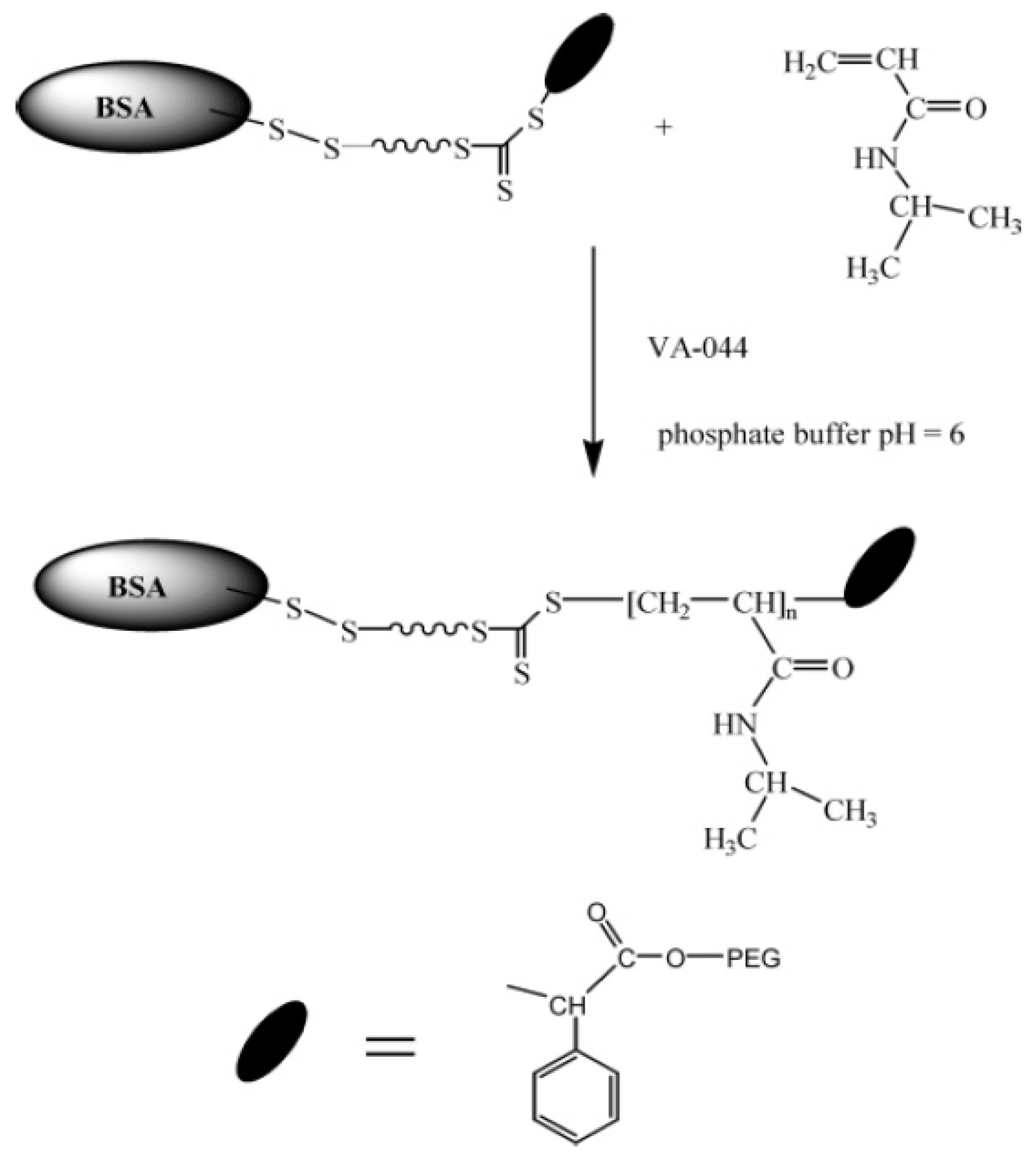
2.1.2. Reversible Addition-Fragmentation Chain Transfer (RAFT)
2.2. Conjugation Methods for the Synthesis of Protein–Polymer Conjugates
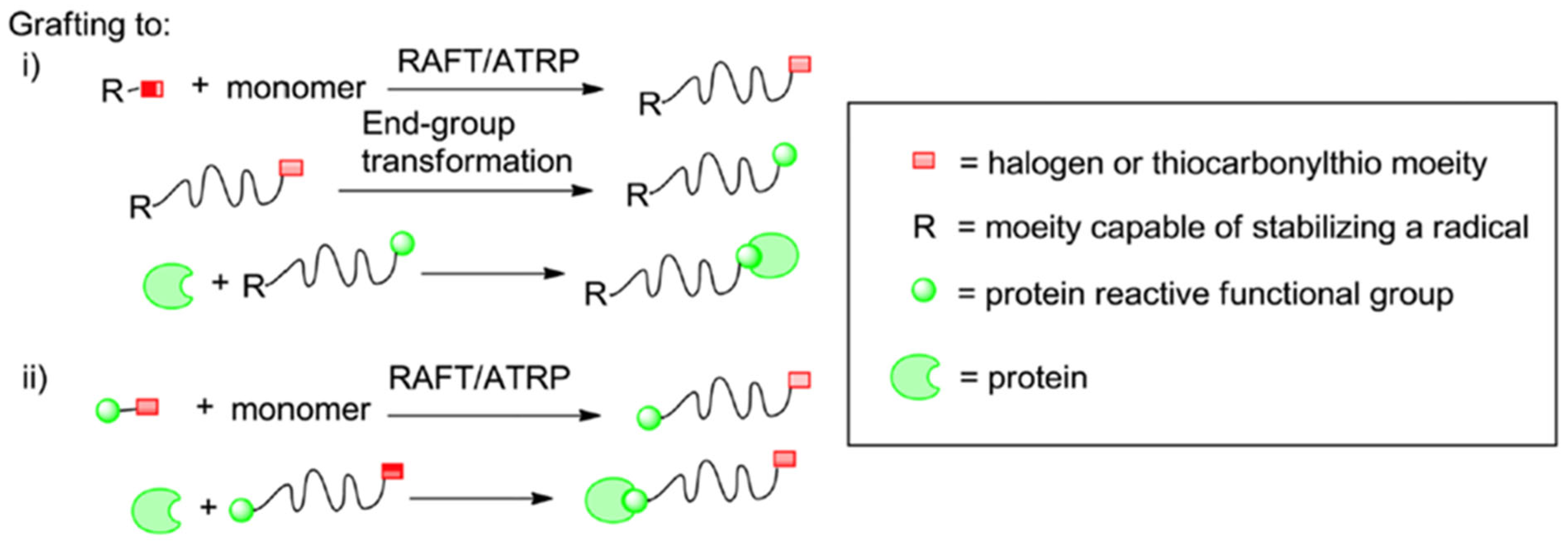
2.2.1. Grafting Polymers to Proteins
- ➢
- For polymers synthesized with ATRP, a nucleophile can replace the halogen attached at the Ω-chain end, thus forming a semi-telechelic polymer apt for conjugation.
- ➢
- Polymers synthesized with RAFT can undergo two different paths of modification targeting the thiocarbonylthio groups as follows:
- Radical coupling with functionalized azoinitiators.
- The use of reducing agents (e.g., Sodium Borohydride) of a nucleophile (e.g., Butylamine) to reduce thiocarbonylthio groups to free thiols [11].
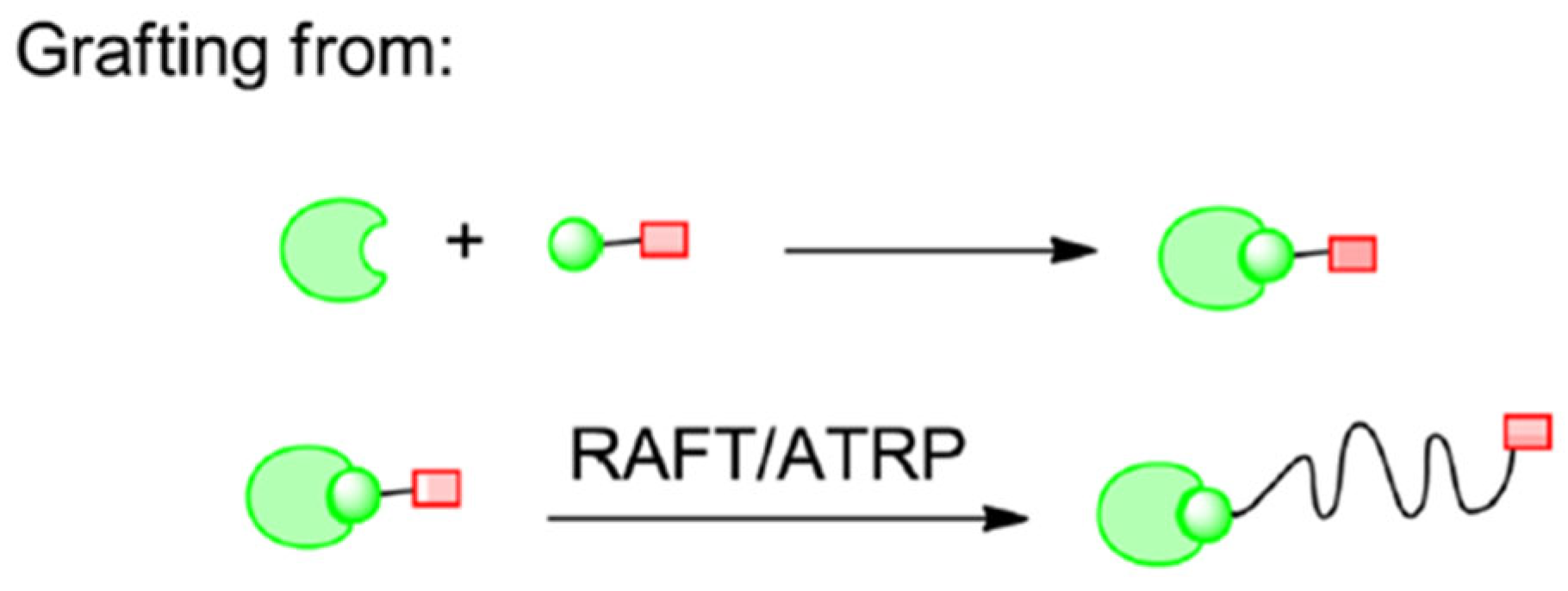
2.2.2. Grafting Polymers from Proteins
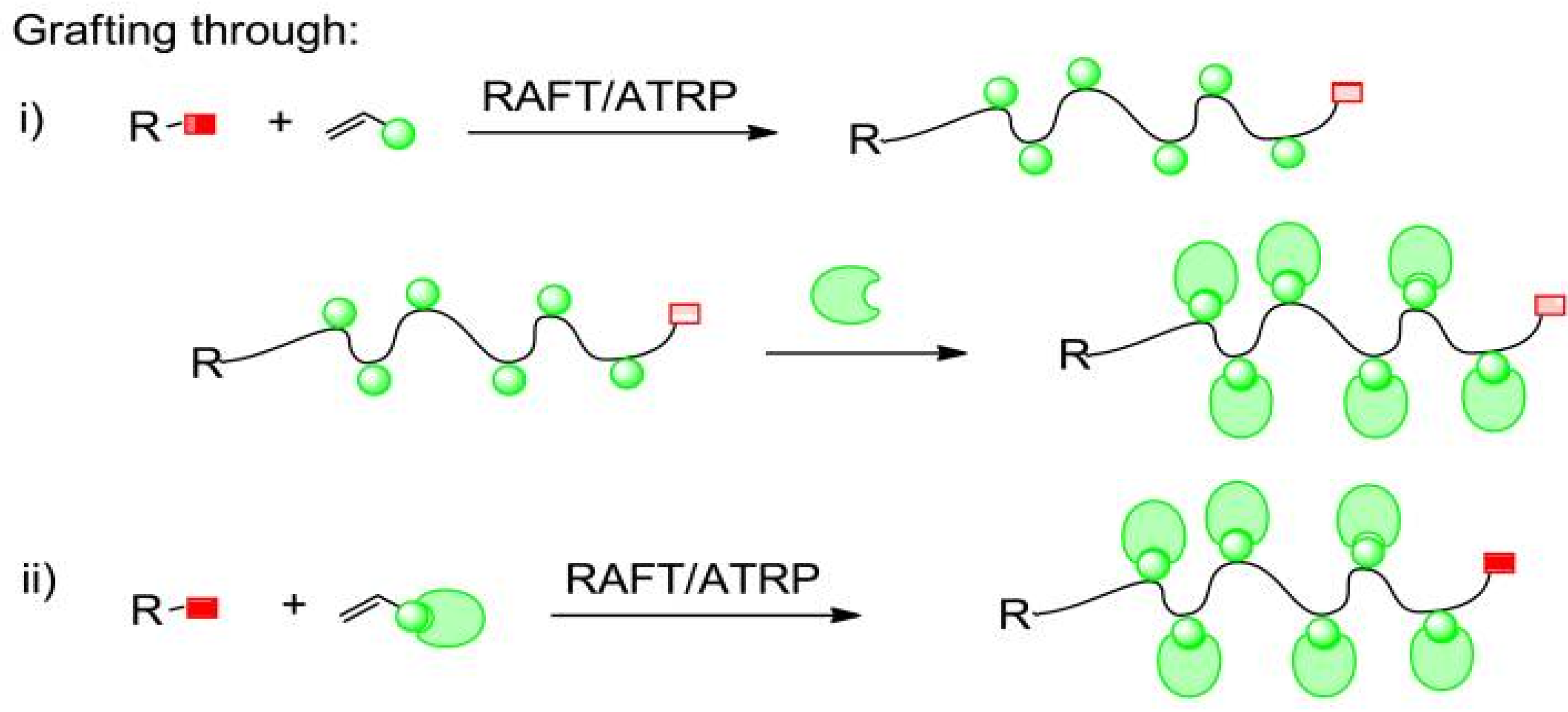
2.2.3. Grafting Through Method
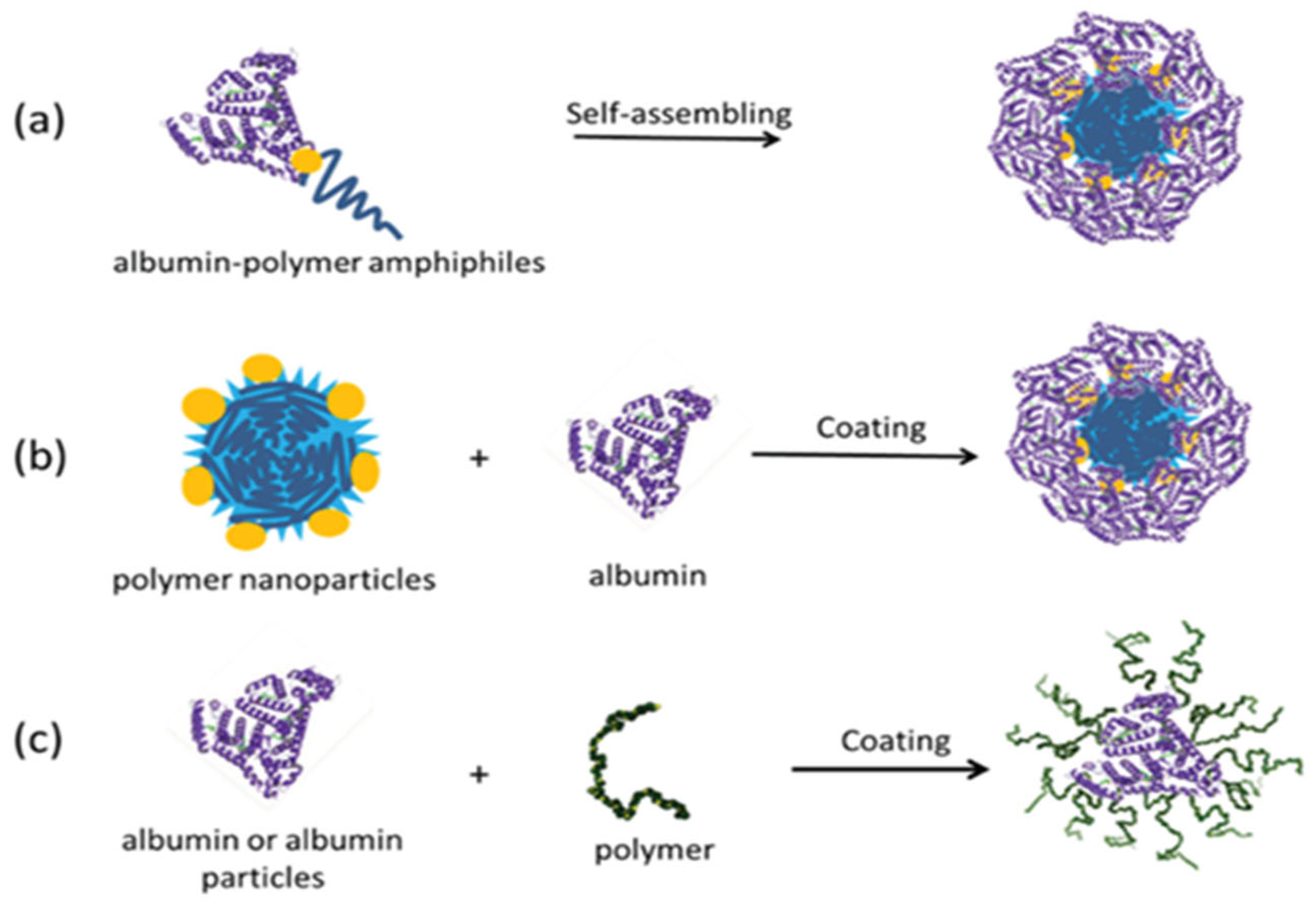
2.3. Albumin–Polymer Conjugates
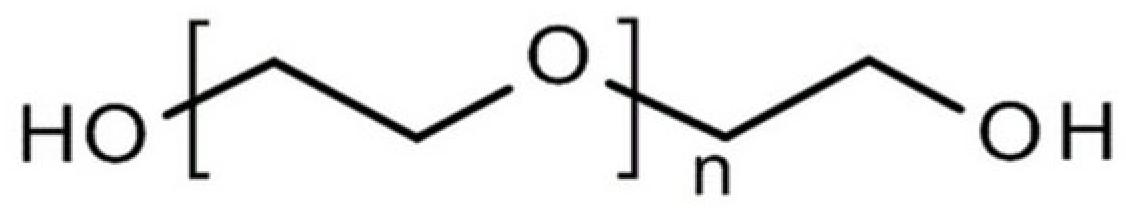
2.4. Polyethylene Glycol (PEG)
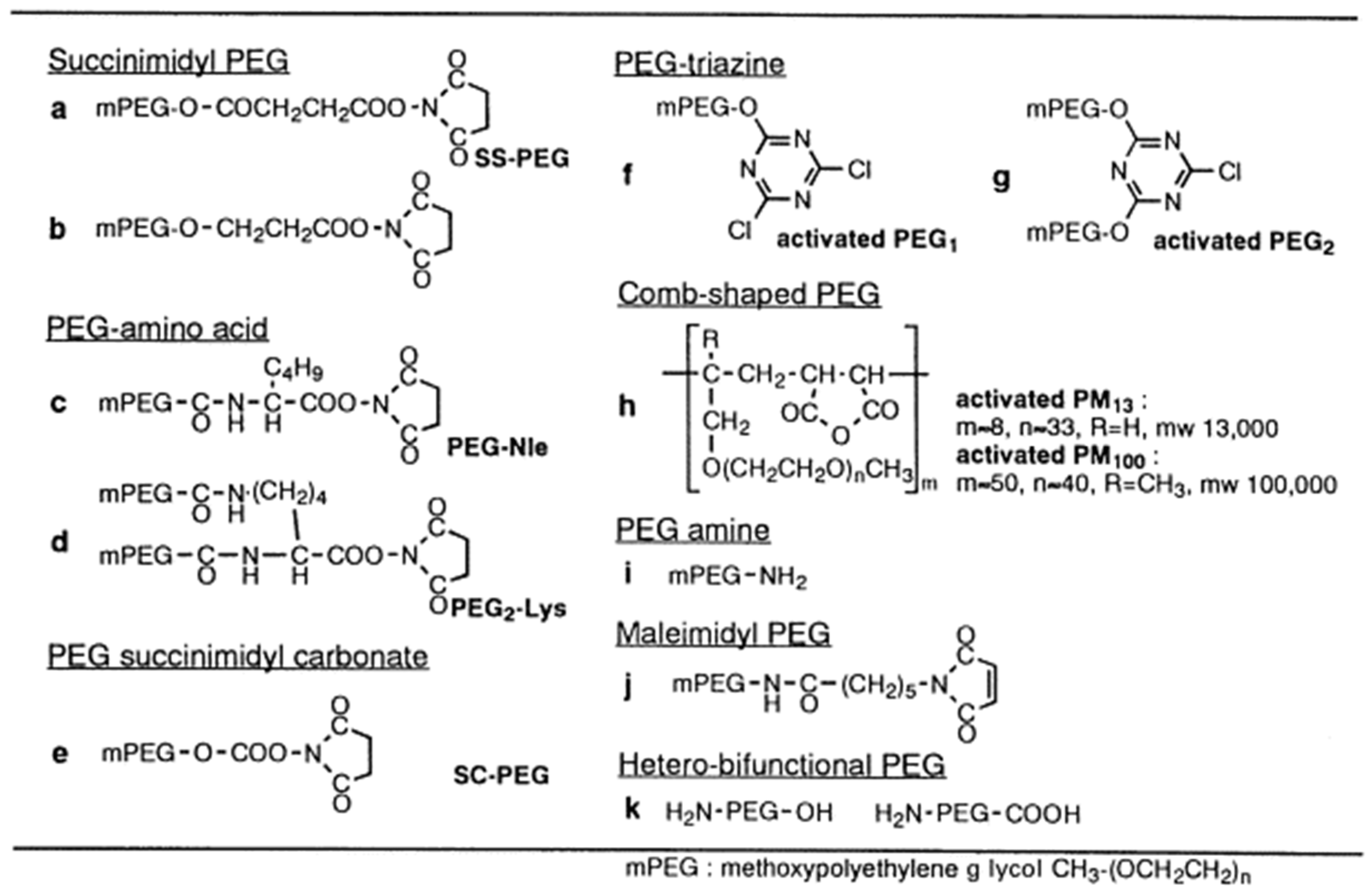
2.4.1. PEG Derivatives
- Succinimidyl PEG
- PEG-succinimidyl carbonate
- PEG-amino acid
- PEG-triazine
- Comb-shaped PEGs
- Other PEG derivatives
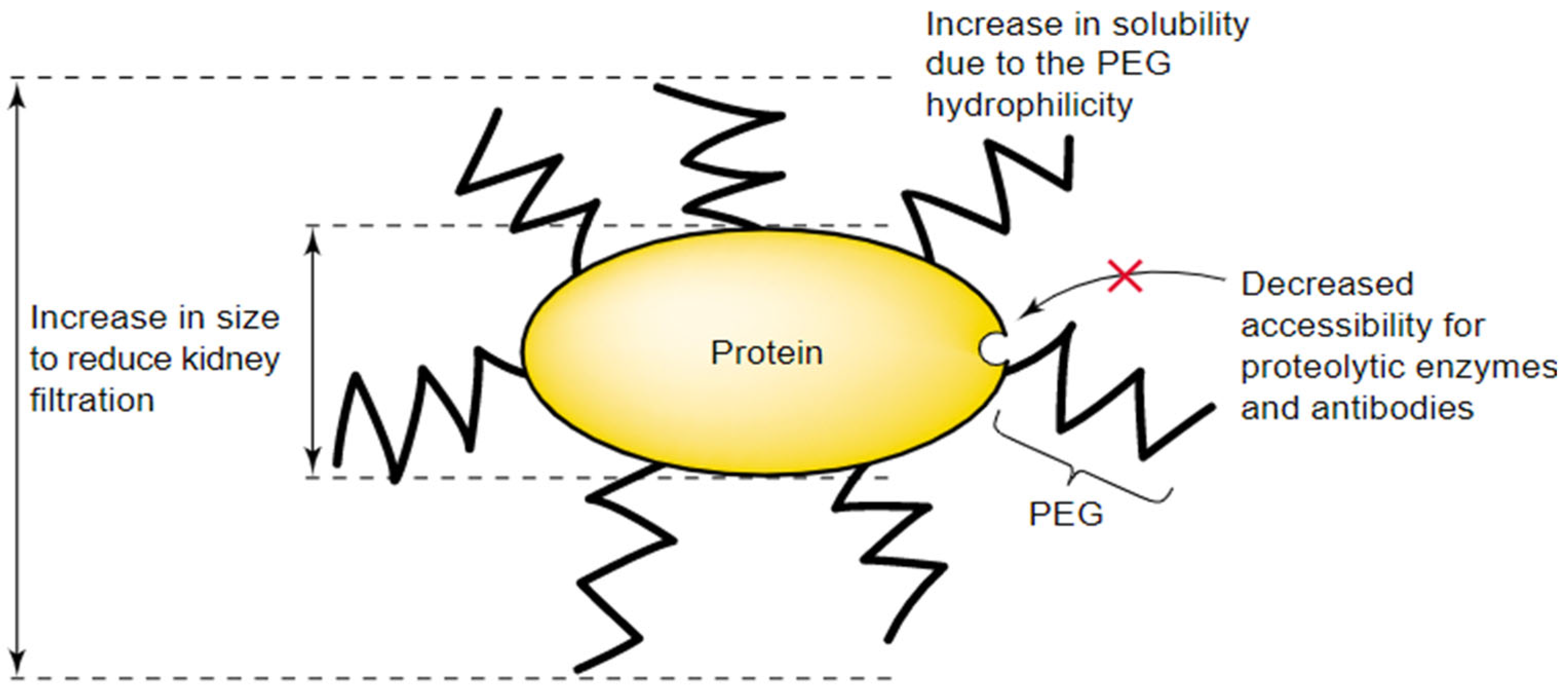
2.4.2. Advantages of PEG-Active Substance Conjugates
2.4.3. Factors Limiting the Use of PEG
2.5. Site-Specific Conjugation
2.6. Protein PEPylation
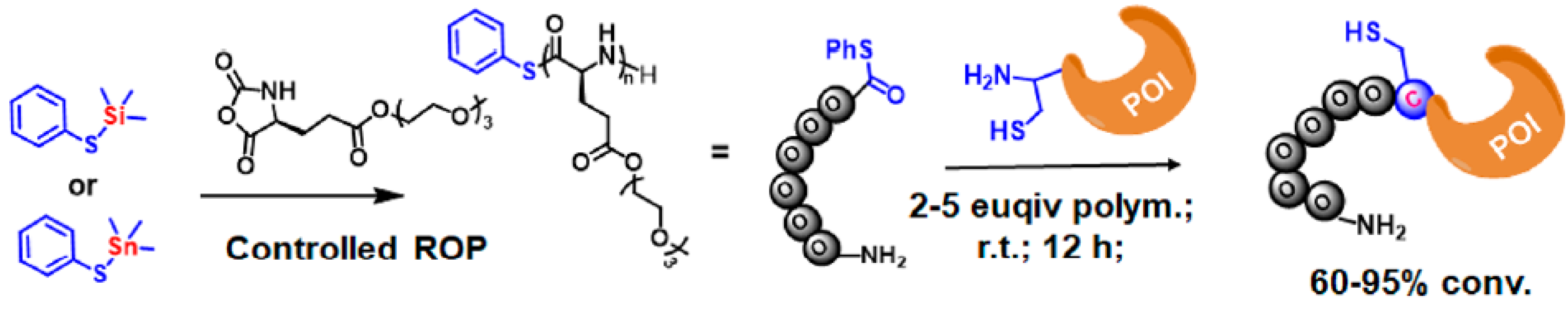
2.6.1. N-Terminal Specific Protein PEPylation via NCL
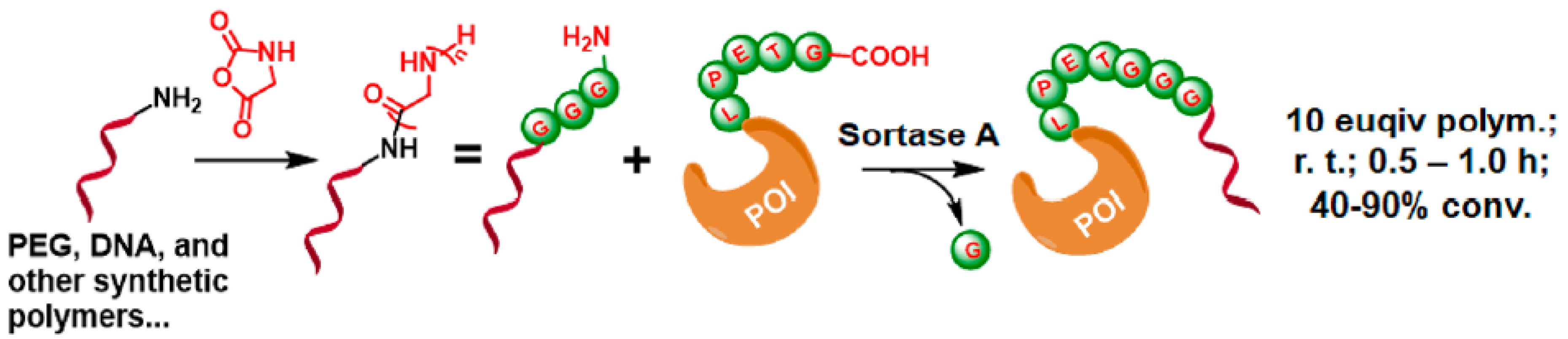
2.6.2. C-Terminal Protein Conjugation via SML (Sortase A-Mediated Ligation)
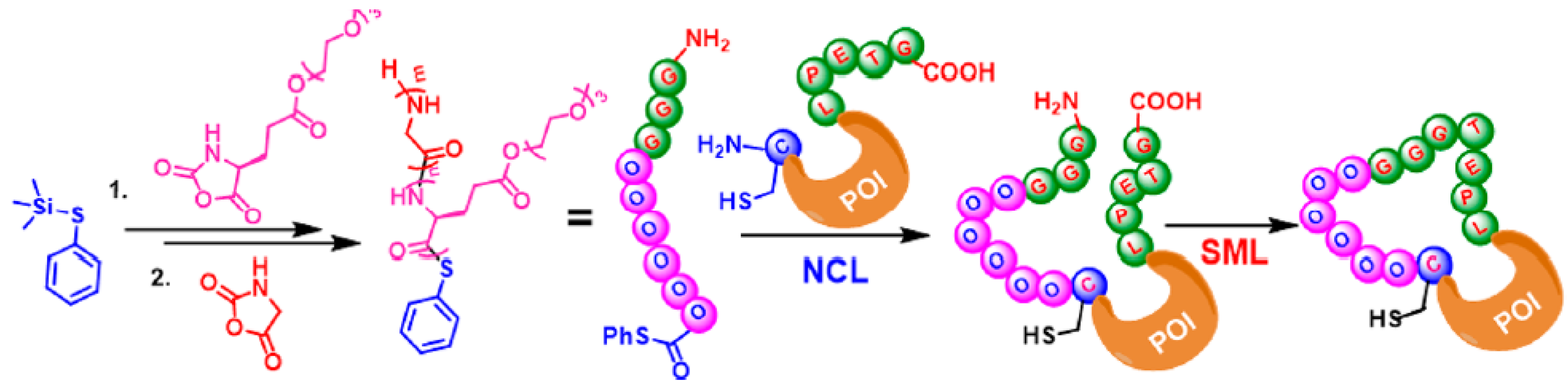
2.6.3. Macrocyclic Protein Conjugation via Consecutive NCL and SML
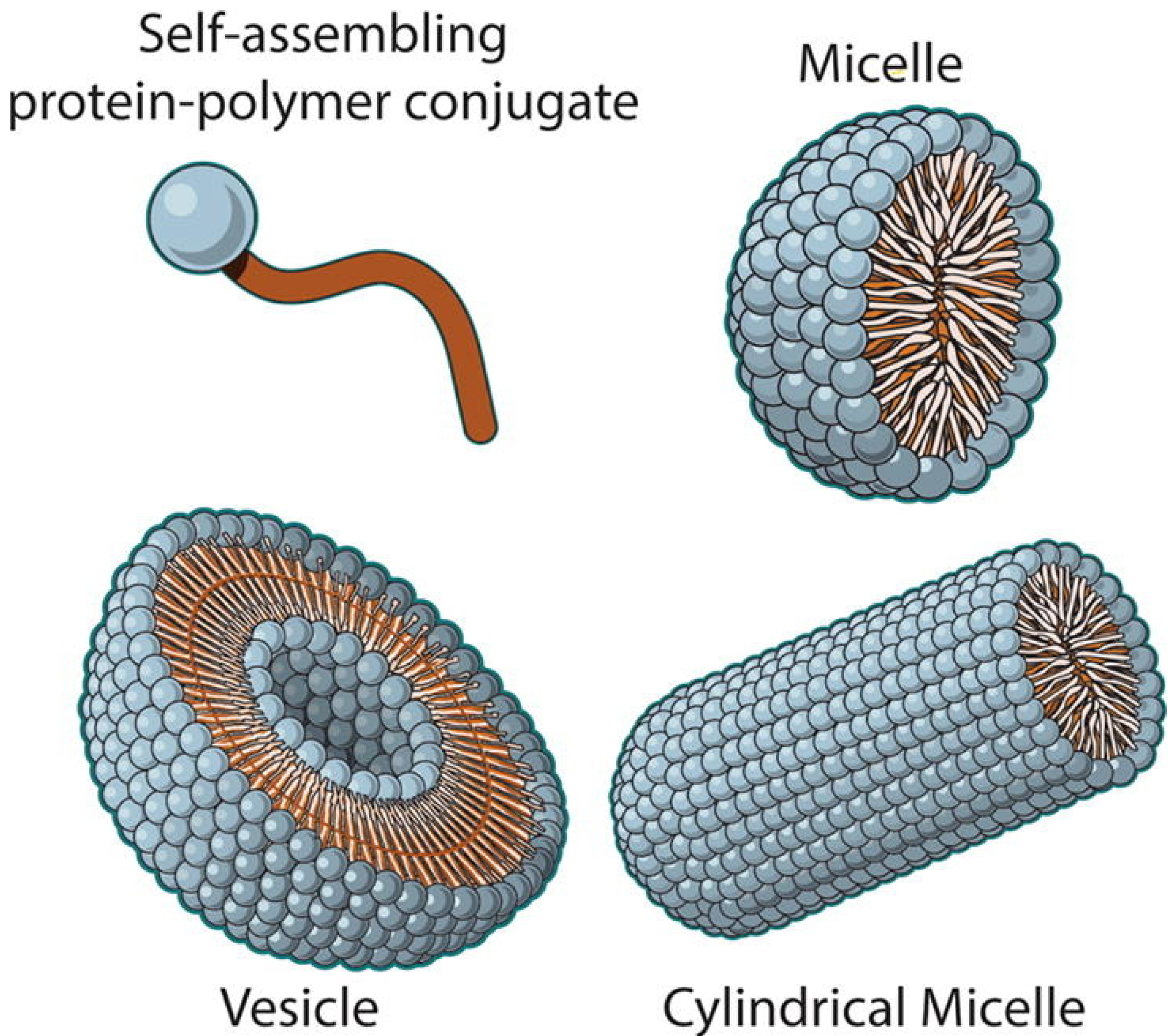
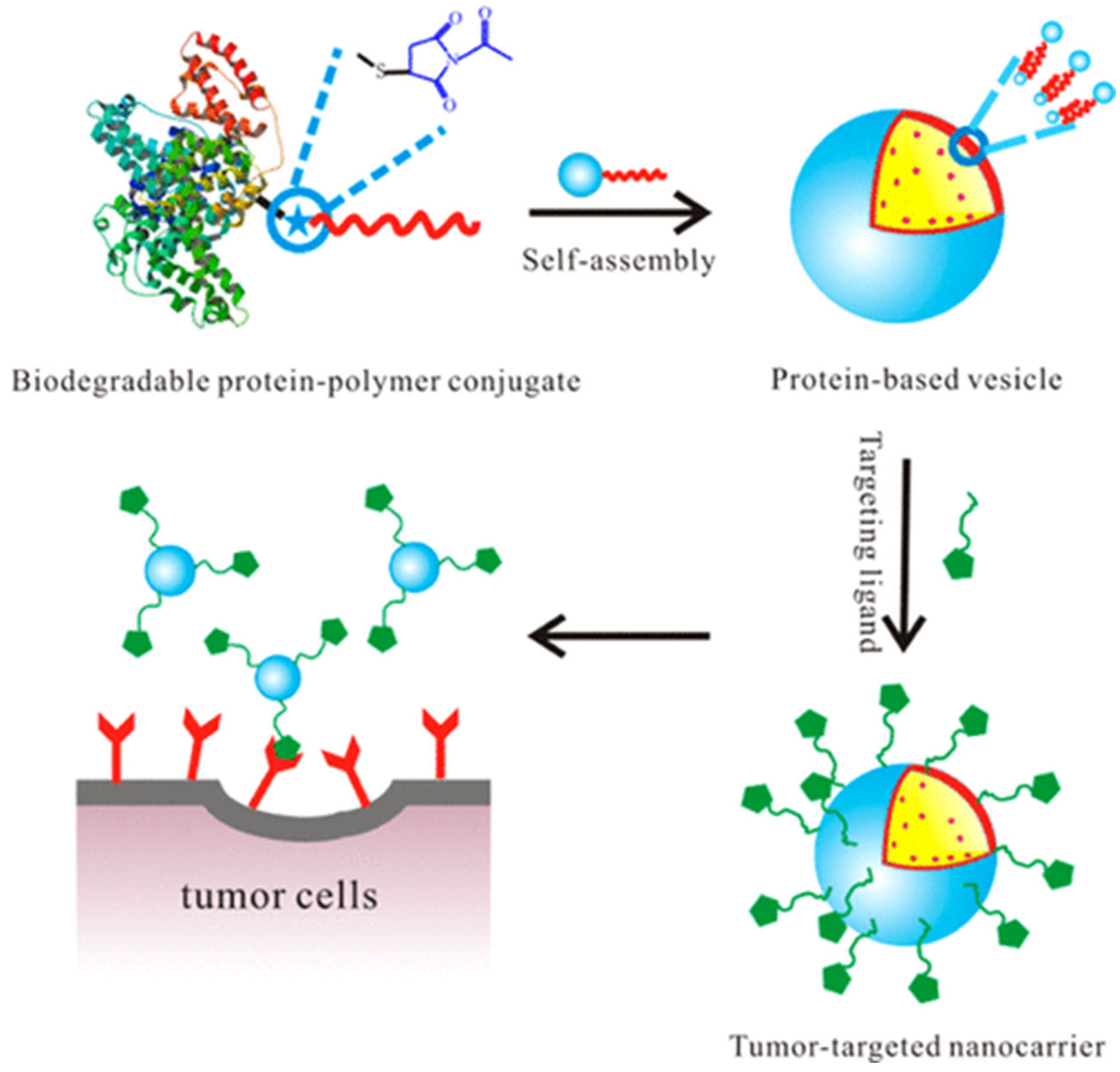
2.7. Nanoparticles Formed with Protein–Polymer Conjugates
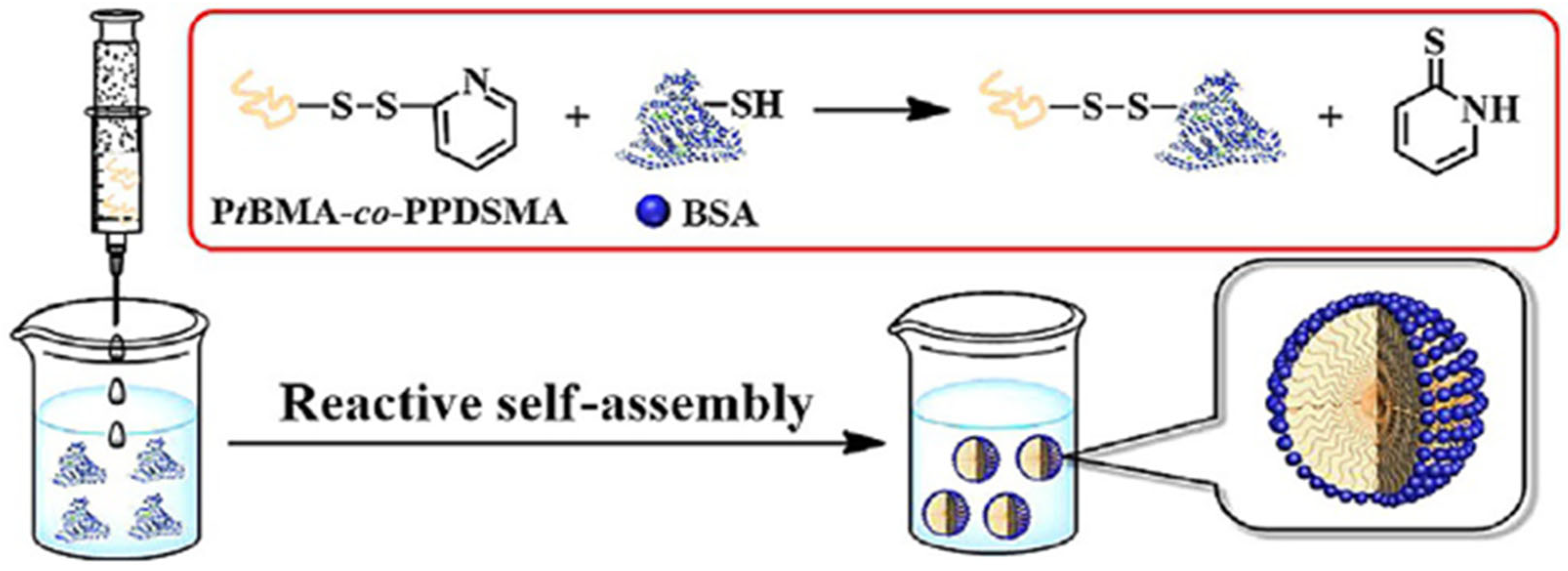
2.8. Synthesis of Bioconjugates Based on Molecular Recognition
- The diversity of proteins and their functional groups makes the design of site-specific bioconjugation quite complex.
- Completing bioconjugation reactions in aqueous media is so difficult due to the hydrophobic nature of the dendron-polymer.
- Conducting chemical analysis or purification processes on this type of hydrophilic bioconjugates is extremely challenging.
- The increase in the concentration of BSA protein results in the formation of smaller CCNs micelles.
- The increase in the concentration of PtBMA-co-PPDSMA polymer results in the formation of bigger CCNs micelles.
- Below LCST: the polymer is extended, and it blocks the conjugation of large biotinylated proteins.
2.9. Synthesis Techniques and Tailored Properties of Protein–Polymer Hydrogels
2.10. Importance of Molecular Weight in Protein–Polymer Conjugates
3. Biodegradability
4. Cytotoxicity
5. Applications
5.1. Biomedical Applications
5.1.1. Drug Delivery
- Temporal control: which is the drug release as a response to an external stimulus (e.g., heat, light, etc.).
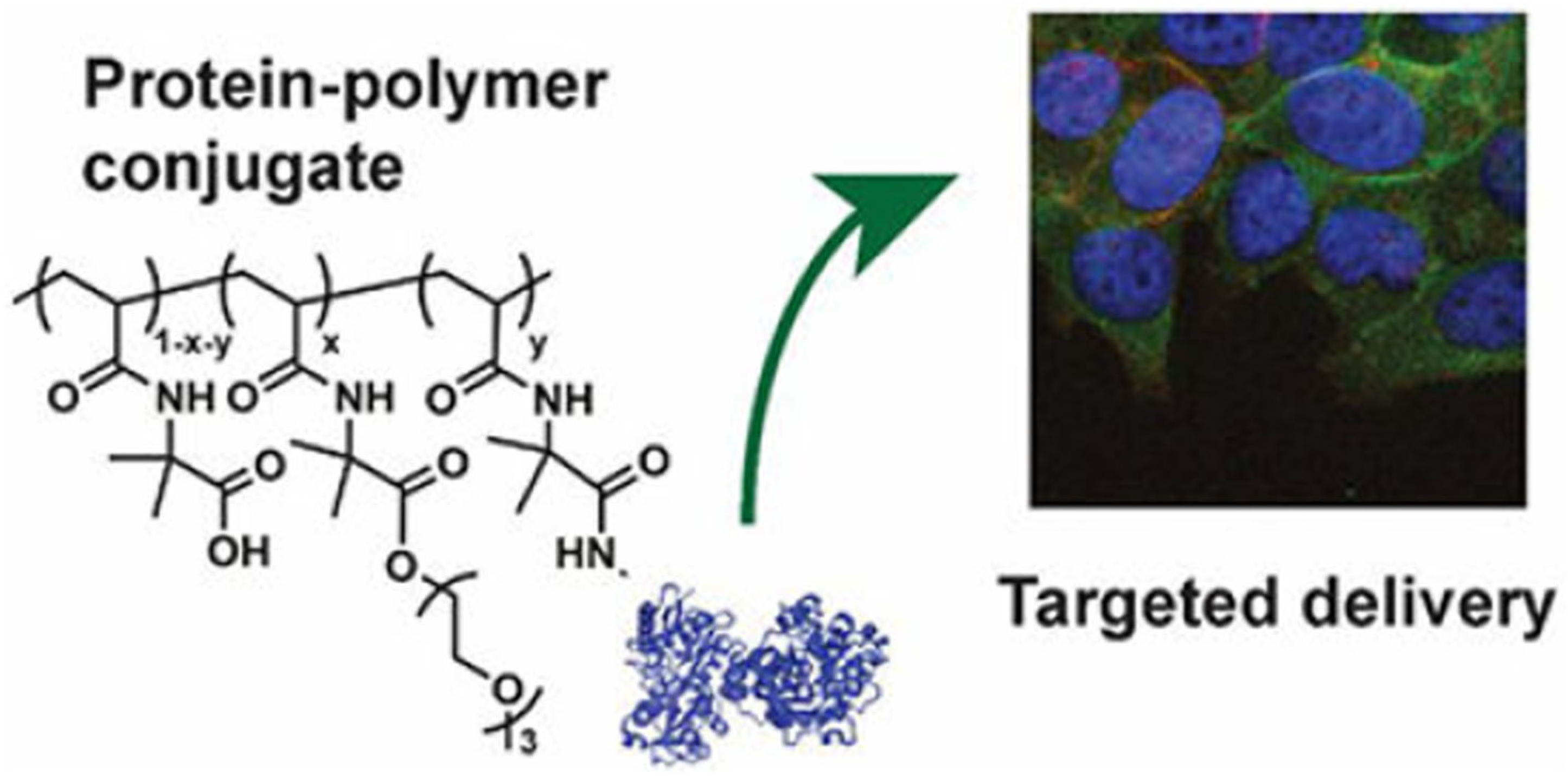
- Distribution control: which is the targeted delivery of the drug [2].
5.1.2. Tissue Engineering
5.1.3. Anti-Cancer/Cancer Therapy Applications
5.2. Miscellaneous Applications
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.A.; Kaur, K.; Klok, H.A. Self-assembly of protein-polymer conjugates for drug delivery. Adv. Drug Deliv. Rev. 2021, 174, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2019, 18, 273–294. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, X.; Li, X.; Ma, C.; Chu, X.; Wang, L.; Xu, W. A review on recent advances of Protein-Polymer hydrogels. Eur. Polym. J. 2022, 162, 110881. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Abbaspourrad, A. Improved pH stability, heat stability, and functionality of phycocyanin after PEGylation. Int. J. Biol. Macromol. 2022, 222, 1758–1767. [Google Scholar] [CrossRef]
- Moulisová, V.; Poveda-Reyes, S.; Sanmartín-Masiá, E.; Quintanilla-Sierra, L.; Salmerón-Sánchez, M.; Ferrer, G.G. Hybrid Protein-Glycosaminoglycan Hydrogels Promote Chondrogenic Stem Cell Differentiation. ACS Omega 2017, 2, 7609–7620. [Google Scholar] [CrossRef]
- Abuchowski, A.; McCoy, J.R.; Palczuk, N.C.; van Es, T.; Davis, F.F. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 1977, 252, 3582–3586. [Google Scholar] [CrossRef]
- Greish, K.; Fang, J.; Inutsuka, T.; Nagamitsu, A.; Maeda, H. Macromolecular Therapeutics: Advantages and Prospects with Special Emphasis on Solid Tumour Targeting. Clin. Pharmacokinet. 2003, 42, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Canalle, L.A.; Löwik, D.W.P.M.; van Hest, J.C.M. Polypeptide–polymer bioconjugates. Chem. Soc. Rev. 2009, 39, 329–353. [Google Scholar] [CrossRef]
- Ju, Y.; Zhang, Y.; Zhao, H. Fabrication of Polymer–Protein Hybrids. Macromol. Rapid Commun. 2018, 39, 1700737. [Google Scholar] [CrossRef]
- Grover, G.N.; Maynard, H.D. Protein-Polymer Conjugates: Synthetic Approaches by Controlled Radical Polymerizations & Interesting Applications. Curr. Opin. Chem. Biol. 2010, 14, 818. [Google Scholar] [CrossRef]
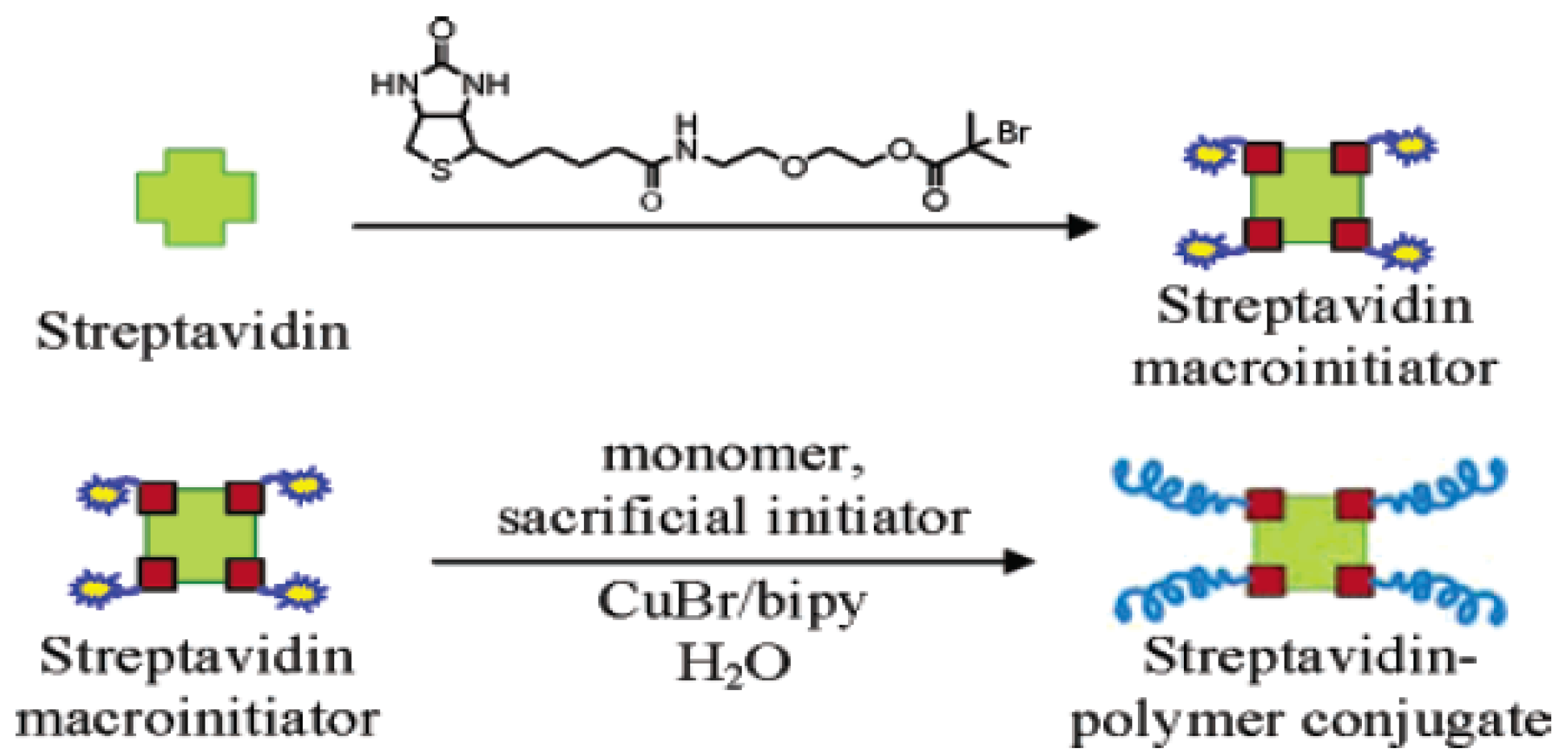
- Bontempo, D.; Maynard, H.D. Streptavidin as a Macroinitiator for Polymerization: In Situ Protein-Polymer Conjugate Formation. J. Am. Chem. Soc. 2005, 127, 6508–6509. [Google Scholar] [CrossRef] [PubMed]
- Boyer, C.; Bulmus, V.; Liu, J.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. Well-defined protein-polymer conjugates via in situ RAFT polymerization. J. Am. Chem. Soc. 2007, 129, 7145–7154. [Google Scholar] [CrossRef]
- Cobo, I.; Li, M.; Sumerlin, B.S.; Perrier, S. Smart hybrid materials by conjugation of responsive polymers to biomacromolecules. Nat. Mater. 2014, 14, 143–159. [Google Scholar] [CrossRef]
- Vega-Hernández, M.Á.; Cano-Díaz, G.S.; Vivaldo-Lima, E.; Rosas-Aburto, A.; Hernández-Luna, M.G.; Martinez, A.; Palacios-Alquisira, J.; Mohammadi, Y.; Penlidis, A. A Review on the Synthesis, Characterization, and Modeling of Polymer Grafting. Processes 2021, 9, 375. [Google Scholar] [CrossRef]
- Jiang, Y.; Stenzel, M. Drug Delivery Vehicles Based on Albumin–Polymer Conjugates. Macromol. Biosci. 2016, 16, 791–802. [Google Scholar] [CrossRef]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Kodera, Y. Pegylation of proteins and bioactive substances for medical and technical applications. Prog. Polym. Sci. 1998, 23, 1233–1271. [Google Scholar] [CrossRef]
- Roberts, M.J.; Bentley, M.D.; Harris, J.M. Chemistry for peptide and protein PEGylation. Adv. Drug Deliv. Rev. 2002, 54, 459–476. [Google Scholar] [CrossRef]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Bontempo, D.; Heredia, K.L.; Fish, B.A.; Maynard, H.D. Cysteine-reactive polymers synthesized by atom transfer radical polymerization for conjugation to proteins. J. Am. Chem. Soc. 2004, 126, 15372–15373. [Google Scholar] [CrossRef]
- Hou, Y.; Lu, H. Protein PEPylation: A New Paradigm of Protein-Polymer Conjugation. Bioconjug. Chem. 2019, 30, 1604–1616. [Google Scholar] [CrossRef]
- An, F.F.; Zhang, X.H. Strategies for Preparing Albumin-based Nanoparticles for Multifunctional Bioimaging and Drug Delivery. Theranostics 2017, 7, 3667–3689. [Google Scholar] [CrossRef]
- Pelegri-O’day, E.M.; Lin, E.-W.; Maynard, H.D. Therapeutic Protein−Polymer Conjugates: Advancing Beyond PEGylation. J. Am. Chem. Soc. 2014, 136, 14323–14332. [Google Scholar] [CrossRef]
- Green, N.M. Avidin. Adv. Protein Chem. 1975, 29, 85–133. [Google Scholar]
- Wang, J.T.; Wang, L.; Ji, X.; Liu, L.; Zhao, H. Synthesis of Zwitterionic Diblock Copolymers with Cleavable Biotin Groups at the Junction Points and Fabrication of Bioconjugates by Biotin−Streptavidin Coupling. Macromolecules 2017, 50, 2284–2295. [Google Scholar] [CrossRef]
- Buller, J.; Laschewsky, A.; Lutz, J.F.; Wischerhoff, E. Tuning the lower critical solution temperature of thermoresponsive polymers by biospecific recognition. Polym. Chem. 2011, 2, 1486–1489. [Google Scholar] [CrossRef]
- Ding, Z.; Fong, R.B.; Long, C.J.; Stayton, P.S.; Hoffman, A.S. Size-dependent control of the binding of biotinylated proteins to streptavidin using a polymer shield. Nature 2001, 411, 59–62. [Google Scholar] [CrossRef]
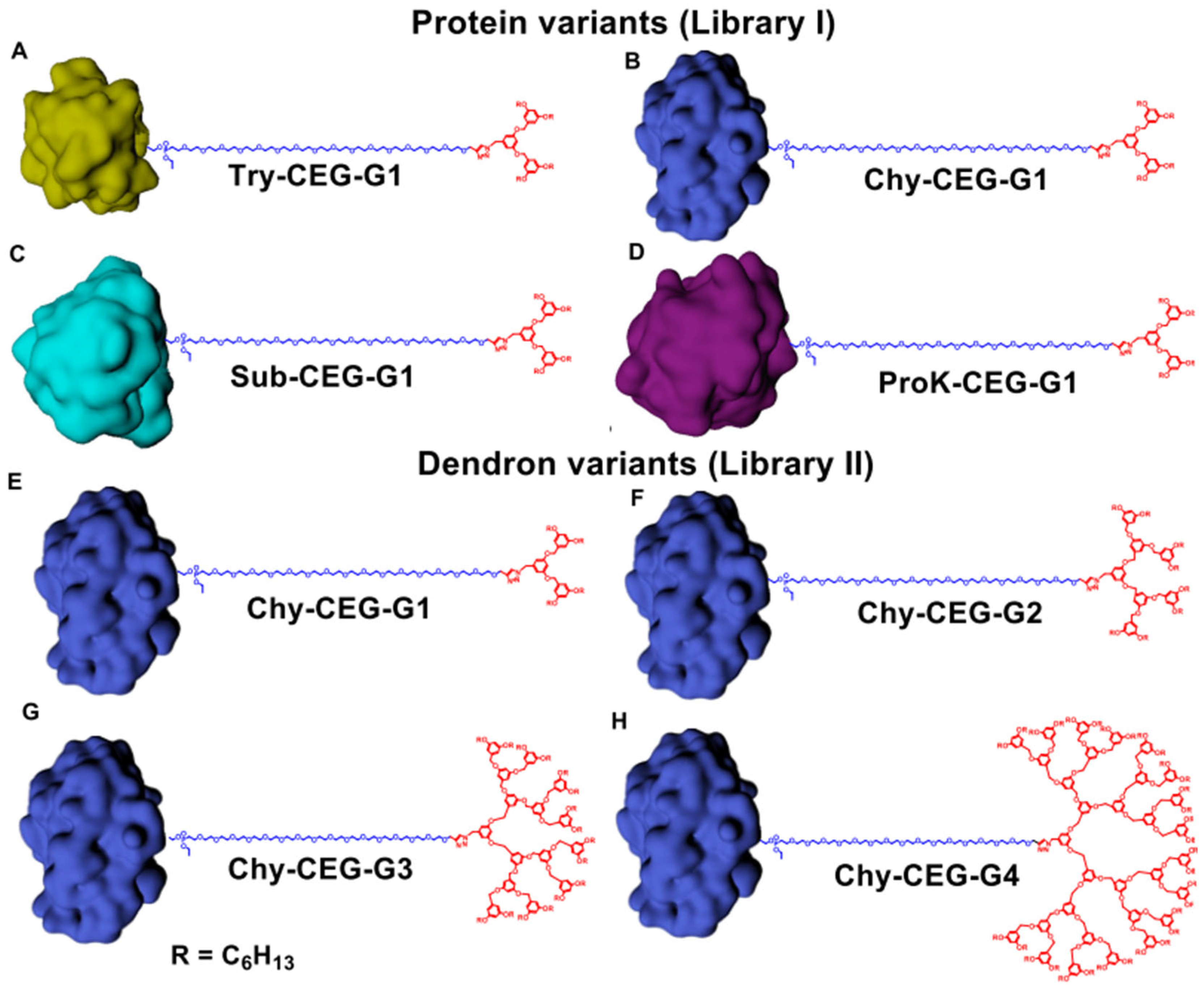
- Sandanaraj, B.S.; Bhandari, P.J.; Reddy, M.M.; Lohote, A.B.; Sahoo, B. Design, Synthesis, and Self-Assembly Studies of a Suite of Monodisperse, Facially Amphiphilic, Protein–Dendron Conjugates. ChemBioChem 2020, 21, 408–416. [Google Scholar] [CrossRef]
- French, A.C.; Thompson, A.L.; Davis, B.G. High-purity discrete PEG-oligomer crystals allow structural insight. Angew. Chem. Int. Ed. 2009, 48, 1248–1252. [Google Scholar] [CrossRef]
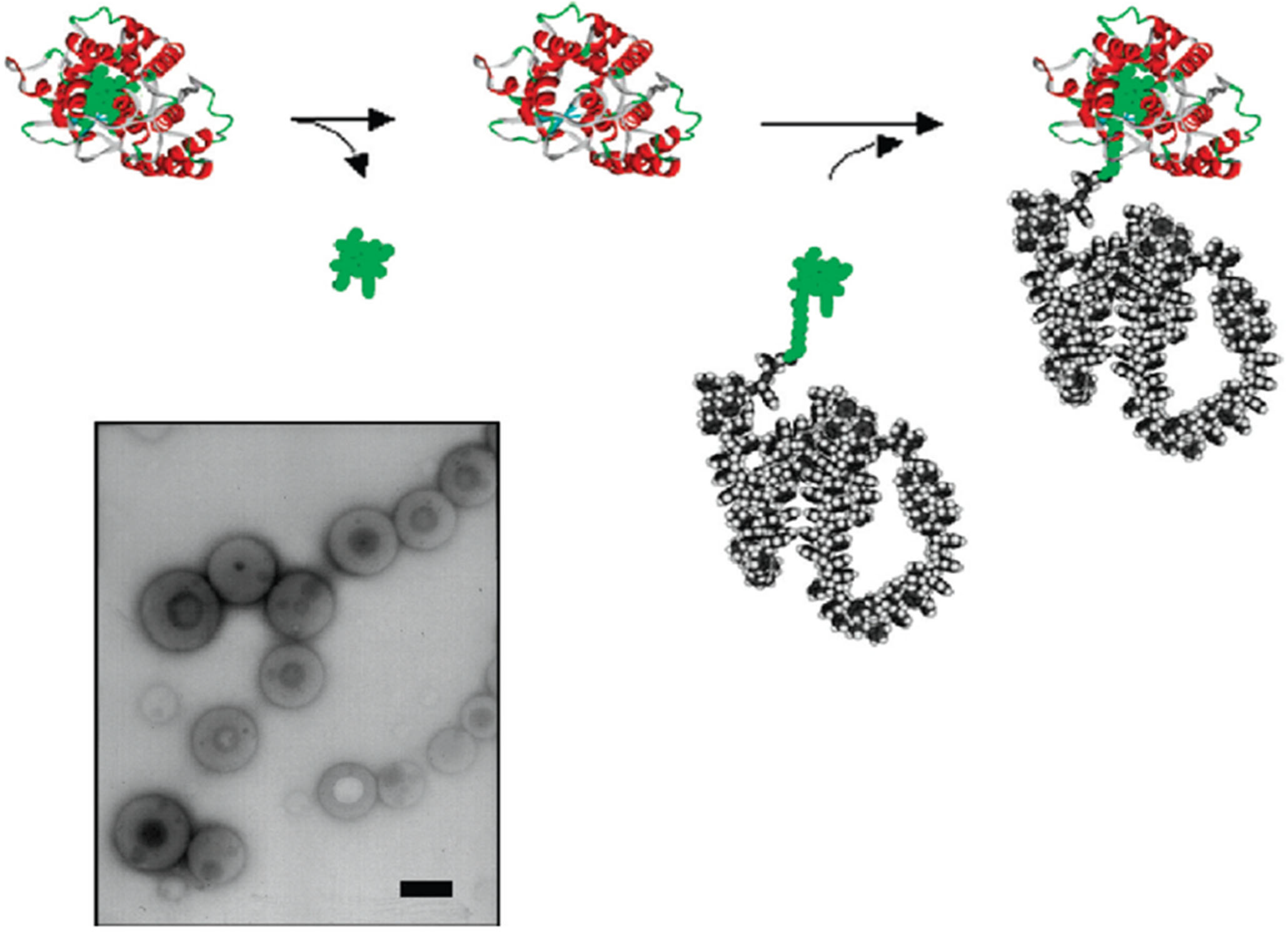
- Ju, Y.; Xing, C.; Wu, D.; Wu, Y.; Wang, L.; Zhao, H. Covalently Connected Polymer–Protein Nanostructures Fabricated by a Reactive Self-Assembly Approach. Chem. Eur. J. 2017, 23, 3366–3374. [Google Scholar] [CrossRef]
- Wang, J.-T.; Hong, Y.; Ji, X.; Zhang, M.; Liu, L.; Zhao, H. In situ fabrication of PHEMA–BSA core–corona biohybrid particles. J. Mater. Chem. B 2016, 4, 4430–4438. [Google Scholar] [CrossRef]
- Lu, L.; Yuan, L.; Yan, J.; Tang, C.; Wang, Q. Development of Core−Shell Nanostructures by In Situ Assembly of Pyridine-Grafted Diblock Copolymer and Transferrin for Drug Delivery Applications. Biomacromolecules 2016, 17, 2321–2328. [Google Scholar] [CrossRef]
- Reynhout, I.C.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Synthesis of Polymer-Biohybrids: From Small to Giant Surfactants. Acc. Chem. Res. 2009, 42, 681–692. [Google Scholar] [CrossRef]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef]
- Pasut, G. Polymers for Protein Conjugation. Polymers 2014, 6, 160–178. [Google Scholar] [CrossRef]
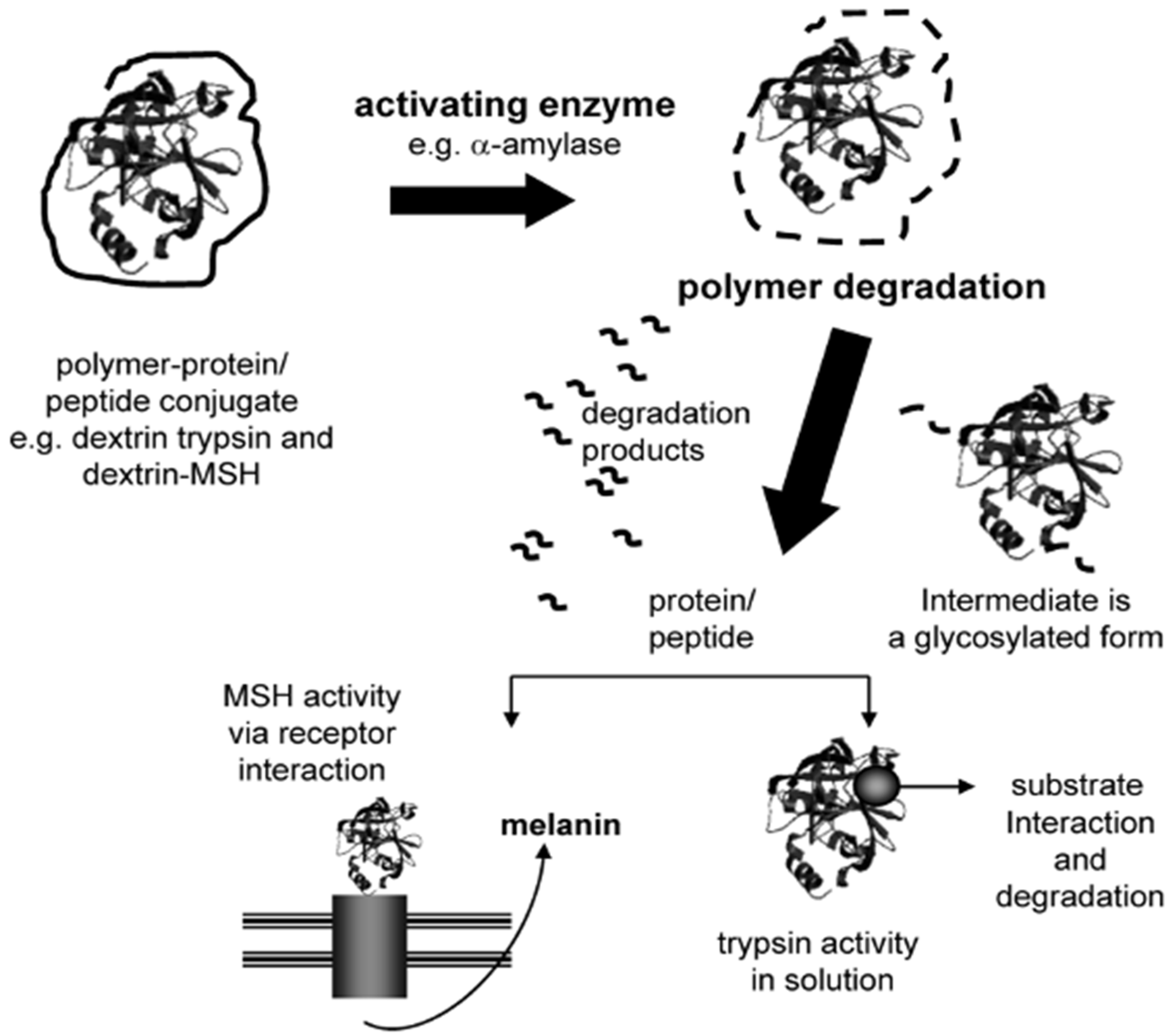
- Duncan, R.; Gilbert, H.R.P.; Carbajo, R.J.; Vicent, M.J. Polymer masked-unmasked protein therapy. 1. Bioresponsive dextrin-trypsin and -melanocyte stimulating hormone conjugates designed for α-amylase activation. Biomacromolecules 2008, 9, 1146–1154. [Google Scholar] [CrossRef]
- Ferguson, E.L.; Alshame, A.M.J.; Thomas, D.W. Evaluation of hyaluronic acid–protein conjugates for polymer masked–unmasked protein therapy. Int. J. Pharm. 2010, 402, 95–102. [Google Scholar] [CrossRef]
- Escalona, G.R.; Sanchis, J.; Vicent, M.J. pH-Responsive Polyacetal-Protein Conjugates Designed for Polymer Masked-Unmasked Protein Therapy (PUMPT). Macromol. Biosci. 2017, 18, 1700302. [Google Scholar] [CrossRef]
- Talelli, M.; Vicent, M.J. Reduction Sensitive Poly(L-glutamic acid) (PGA)-Protein Conjugates Designed for Polymer Masked−Unmasked Protein Therapy. Biomacromolecules 2014, 15, 4168–4177. [Google Scholar] [CrossRef]
- Greco, F.; Arif, I.; Botting, R.; Fante, C.; Quintieri, L.; Clementi, C.; Schiavon, O.; Pasut, G. Polysialic acid as a drug carrier: Evaluation of a new polysialic acid-epirubicin conjugate and its comparison against established drug carriers. Polym. Chem. 2013, 4, 1600–1609. [Google Scholar] [CrossRef]
- Saman, H.; Saman, H.; Uddin, S.; Raza, S.; Shrimali, R.; Rasul, K. Understanding Checkpoint Inhibitors in Cancer Therapy, Mechanisms of Action, Resistance and Future Challenges. Clin. Oncol. Res. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Fan, F.; Wood, K.V. Bioluminescent Assays for High-Throughput Screening. ASSAY Drug Dev. Technol. 2007, 5, 127–136. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A. Use of Multiple Assay Endpoints to Investigate the Effects of Incubation Time, Dose of Toxin, Plating Density in Cell-Based Cytotoxicity Assays. ASSAY Drug Dev. Technol. 2004, 2, 51–62. [Google Scholar] [CrossRef]
- Spicer, C.D.; Jumeaux, C.; Gupta, B.; Stevens, M.M. Peptide and protein nanoparticle conjugates: Versatile platforms for biomedical applications. Chem. Soc. Rev. 2018, 47, 3574. [Google Scholar] [CrossRef]
- Priestman, T.J. Cancer Chemotherapy: An Introduction; Springer: Dordrecht, The Netherlands, 1989. [Google Scholar] [CrossRef]
- Decker, T.; Lohmann-Matthes, M.L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 1988, 115, 61–69. [Google Scholar] [CrossRef]
- Niles, A.L.; Moravec, R.A.; Hesselberth, P.E.; Scurria, M.A.; Daily, W.J.; Riss, T.L. A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers. Anal. Biochem. 2007, 366, 197–206. [Google Scholar] [CrossRef]
- Dearden, J.C. In silico prediction of drug toxicity. J. Comput. Aided Mol. Des. 2003, 17, 119–127. [Google Scholar] [CrossRef]
- Gavanji, S.; Bakhtari, A.; Famurewa, A.C.; Othman, E.M. Cytotoxic Activity of Herbal Medicines as Assessed in Vitro: A Review. Chem. Biodivers. 2023, 20, e202201098. [Google Scholar] [CrossRef]
- Qi, Y.; Amiram, M.; Gao, W.; McCafferty, D.G.; Chilkoti, A. Sortase-Catalyzed Initiator Attachment Enables High Yield Growth of a Stealth Polymer from the C Terminus of a Protein. Macromol. Rapid Commun. 2013, 34, 1256–1260. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Liu, W.; Wang, W.; Weitzhandler, I.; Li, X.; Qi, Y.; Liu, J.; Pang, Y.; Hunt, D.F.; Chilkoti, A. Site-Specific Zwitterionic Polymer Conjugates of a Protein Have Long Plasma Circulation. ChemBioChem 2015, 16, 2451–2455. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.J.; Baker, S.L.; Colina, C.M.; Figg, C.A.; Kaar, J.L.; Matyjaszewski, K.; Simakova, A.; Sumerlin, B.S. Next generation protein-polymer conjugates. AIChE J. 2018, 64, 3230–3245. [Google Scholar] [CrossRef]
- Wu, Y.; Ng, D.Y.W.; Kuan, S.L.; Weil, T. Protein-polymer therapeutics: A macromolecular perspective. Biomater. Sci. 2015, 3, 214–230. [Google Scholar] [CrossRef]
- Schellekens, H.; Hennink, W.E.; Brinks, V. The immunogenicity of polyethylene glycol: Facts and fiction. Pharm. Res. 2013, 30, 1729–1734. [Google Scholar] [CrossRef]
- Soares, D.C.F.; Oda, C.M.R.; Monteiro, L.O.F.; de Barros, A.L.B.; Tebaldi, M.L. Responsive polymer conjugates for drug delivery applications: Recent advances in bioconjugation methodologies. J. Drug Target. 2019, 27, 355–366. [Google Scholar] [CrossRef]
- Fee, C.J. Protein conjugates purification and characterization. In PEGylated Protein Drugs: Basic Science and Clinical Applications; Birkhäuser: Basel, Switzerland, 2009. [Google Scholar] [CrossRef]
- Majoros, I.J.; Thomas, T.P.; Mehta, C.B.; Baker, J.R. Poly(amidoamine) dendrimer-based multifunctional engineered nanodevice for cancer therapy. J. Med. Chem. 2005, 48, 5892–5899. [Google Scholar] [CrossRef]
- Seidi, F.; Jenjob, R.; Crespy, D. Designing Smart Polymer Conjugates for Controlled Release of Payloads. Chem. Rev. 2018, 118, 3965–4036. [Google Scholar] [CrossRef]
- Vanparijs, N.; De Coen, R.; Laplace, D.; Louage, B.; Maji, S.; Lybaert, L.; Hoogenboom, R.; De Geest, B.G. Transiently responsive protein-polymer conjugates via a grafting-from RAFT approach for intracellular co-delivery of proteins and immune-modulators. Chem. Commun. 2015, 51, 13972–13975. [Google Scholar] [CrossRef]
- Zou, Y.; Brooks, D.E.; Kizhakkedathu, J.N. A novel functional polymer with tunable LCST. Macromolecules 2008, 41, 5393–5405. [Google Scholar] [CrossRef]
- Wong, C.K.; Laos, A.J.; Soeriyadi, A.H.; Wiedenmann, J.; Curmi, P.M.G.; Gooding, J.J.; Marquis, C.P.; Stenzel, M.H.; Thordarson, P. Polymersomes prepared from thermoresponsive fluorescent protein-polymer bioconjugates: Capture of and report on drug and protein payloads. Angew. Chem. Int. Ed. 2015, 54, 5317–5322. [Google Scholar] [CrossRef]
- Ji, X.; Liu, L.; Zhao, H. Synthesis and self-assembly of bioconjugates composed of thermal-responsive polymer chains and pendant lysozyme molecules. Polym. Chem. 2017, 8, 2815–2823. [Google Scholar] [CrossRef]
- Heredia, K.L.; Bontempo, D.; Ly, T.; Byers, J.T.; Halstenberg, S.; Maynard, H.D. In Situ Preparation of Protein-“Smart” Polymer Conjugates with Retention of Bioactivity. J. Am. Chem. Soc. 2005, 127, 16955–16960. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Morbidelli, M. Process for protein PEGylation. J. Contr. Rel. 2014, 180, 134–149. [Google Scholar] [CrossRef]
- Yang, B.; Lim, S.I.; Kim, J.C.; Tae, G.; Kwon, I. Site-Specific Albumination as an Alternative to PEGylation for the Enhanced Serum Half-Life in Vivo. Biomacromolecules 2016, 17, 1811–1817. [Google Scholar] [CrossRef]
- Gil, M.S.; Cho, J.; Thambi, T.; Phan, V.H.G.; Kwon, I.; Lee, D.S. Bioengineered robust hybrid hydrogels enrich the stability and efficacy of biological drugs. J. Control. Release 2017, 267, 119–132. [Google Scholar] [CrossRef]
- Gao, Y.; Kieltyka, R.E.; Jesse, W.; Norder, B.; Korobko, A.V.; Kros, A. Thiolated human serum albumin cross-linked dextran hydrogels as a macroscale delivery system. Soft Matter 2014, 10, 4869–4874. [Google Scholar] [CrossRef]
- Noteborn, W.E.M.; Gao, Y.; Jesse, W.; Kros, A.; Kieltyka, R.E. Dual-Crosslinked Human Serum Albumin-Polymer Hydrogels for Affinity-Based Drug Delivery. Macromol. Mater. Eng. 2017, 302, 1700243. [Google Scholar] [CrossRef]
- Kim, I.; Choi, J.S.; Lee, S.; Byeon, H.J.; Lee, E.S.; Shin, B.S.; Choi, H.-G.; Lee, K.C.; Youn, Y.S. In situ facile-forming PEG cross-linked albumin hydrogels loaded with an apoptotic TRAIL protein. J. Control. Release 2015, 214, 30–39. [Google Scholar] [CrossRef]
- Phan, V.G.; Duong, H.T.T.; Thambi, T.; Nguyen, T.L.; Turabee, H.; Yin, Y.; Kim, S.H.; Kim, J.; Jeong, J.H.; Lee, D.S. Modularly engineered injectable hybrid hydrogels based on protein-polymer network as potent immunologic adjuvant in vivo. Biomaterials 2019, 195, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liang, M.; Svejkar, D.; Hart-Smith, G.; Lu, H.; Scarano, W.; Stenzel, M.H. Albumin-micelles via a one-pot technology platform for the delivery of drugs. Chem. Commun. 2014, 50, 6394–6397. [Google Scholar] [CrossRef]
- Kim, J.S.; Sirois, A.R.; Cegla, A.J.V.; Jumai’an, E.; Murata, N.; Buck, M.E.; Moore, S.J. Protein-Polymer Conjugates Synthesized Using Water-Soluble Azlactone-Functionalized Polymers Enable Receptor-Specific Cellular Uptake toward Targeted Drug Delivery. Bioconjug. Chem. 2019, 30, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Gonen-Wadmany, M.; Oss-Ronen, L.; Seliktar, D. Protein-polymer conjugates for forming photopolymerizable biomimetic hydrogels for tissue engineering. Biomaterials 2007, 28, 3876–3886. [Google Scholar] [CrossRef]
- Gonen-Wadmany, M.; Oss-Ronen, L.; Seliktar, D. Covalently Adaptable Elastin-Like Protein–Hyaluronic Acid (ELP–HA) Hybrid Hydrogels with Secondary Thermoresponsive Crosslinking for Injectable Stem Cell Delivery. Adv. Funct. Mater. 2017, 27, 1605609. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, H.; Trinh, P.; Heilshorn, S.C.; Yang, F. Elastin-like protein-hyaluronic acid (ELP-HA) hydrogels with decoupled mechanical and biochemical cues for cartilage regeneration. Biomaterials 2017, 127, 132–140. [Google Scholar] [CrossRef] [PubMed]
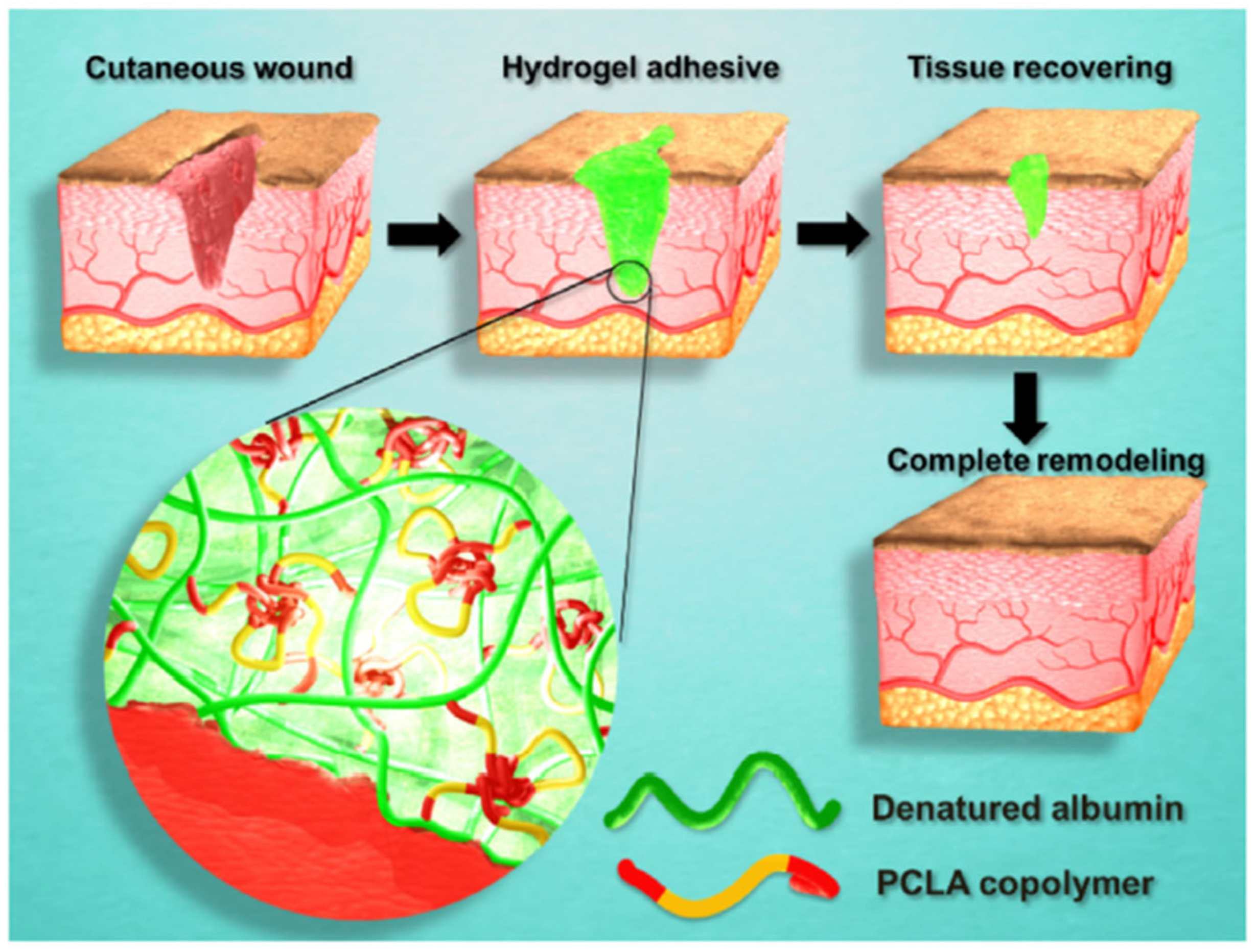
- Phan, V.H.G.; Le, T.M.D.; Janarthanan, G.; Ngo, P.-K.T.; Lee, D.S.; Thambi, T. Development of bioresorbable smart injectable hydrogels based on thermo-responsive copolymer integrated bovine serum albumin bioconjugates for accelerated healing of excisional wounds. J. Ind. Eng. Chem. 2021, 96, 345–355. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, J.; Meng, X.; Gong, C.; Wu, P.; Yang, Z.; Dong, H. ROS-Activated nanoscale coordination polymers for enhanced ultrasound-mediated therapy for the treatment of cancer. Acta Biomater. 2022, 143, 372–380. [Google Scholar] [CrossRef]
- Bagheri, N.; Lakouraj, M.M.; Seyed, A.; Nabavi, R.; Tashakkorian, H.; Mohseni, M. Synthesis of bioactive polyaniline-b-polyacrylic acid copolymer nanofibrils as an effective antibacterial and anticancer agent in cancer therapy, especially for HT29 treatment. RSC Adv. 2020, 10, 25290–25304. [Google Scholar] [CrossRef]
- Takahashi, H.; Yumoto, K.; Yasuhara, K.; Nadres, E.T.; Kikuchi, Y.; Buttitta, L.; Taichman, R.S.; Kuroda, K. Anticancer polymers designed for killing dormant prostate cancer cells. Sci. Rep. 2019, 9, 1096. [Google Scholar] [CrossRef]
- Shao, N.; Yuan, L.; Ma, P.; Zhou, M.; Xiao, X.; Cong, Z.; Wu, Y.; Xiao, G.; Fei, J.; Liu, R. Heterochiral β-Peptide Polymers Combating Multidrug-Resistant Cancers Effectively without Inducing Drug Resistance. J. Am. Chem. Soc. 2022, 144, 7283–7294. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.K.; Thotakura, N.; Kumar, R.; Kumar, P.; Singh, B.; Chitkara, D.; Raza, K. Chitosan-modified PLGA polymeric nanocarriers with better delivery potential for tamoxifen. Int. J. Biol. Macromol. 2016, 93, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-S.; Li, Z.-Y.; Zhu, J.-Y.; Han, K.; Zeng, Z.-Y.; Hong, W.; Li, W.-X.; Jia, H.-Z.; Liu, Y.; Zhuo, R.-X.; et al. Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small 2015, 11, 2543–2554. [Google Scholar] [CrossRef]
- Shakya, A.K.; Sami, H.; Srivastava, A.; Kumar, A. Stability of responsive polymer-protein bioconjugates. Prog. Polym. Sci. 2010, 35, 459–486. [Google Scholar] [CrossRef]
- Huang, A.; Qin, G.; Olsen, B.D. Highly active biocatalytic coatings from protein-polymer diblock copolymers. ACS Appl. Mater. Interfaces 2015, 7, 14660–14669. [Google Scholar] [CrossRef]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic acid in the third millennium. Polymers 2018, 10, 701. [Google Scholar] [CrossRef]
- Castelltort, M.R.; Jimenez, I.O.; Sastre, J.Q. Conjugate of Hyaluronic acid for Cosmetic Treatment and Preparation Method. U.S. Patent US8901080B2, 2 December 2014. [Google Scholar]
- Cho, K.Y.; Moon, J.Y.; Lee, Y.W.; Lee, K.G.; Yeo, J.H.; Kweon, H.Y.; Kim, K.H.; Cho, C.S. Preparation of self-assembled silk sericin nanoparticles. Int. J. Biol. Macromol. 2003, 32, 36–42. [Google Scholar] [CrossRef]
- Kumar, J.P.; Mandal, B.B. The inhibitory effect of silk sericin against ultraviolet-induced melanogenesis and its potential use in cosmeceutics as an anti-hyperpigmentation compound. Photochem. Photobiol. Sci. 2019, 18, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Choung, E.S.; Eom, S.Y.; Kim, J.H.; Kim, K.S.; Kim, K.H.; Lee, K.G.; Lee, Y.W.; Cho, C.S. Synthesis, Characterization and Cosmetic Application of Self-Assembled Sericin-PEG Nanoparticle. In Proceedings of the SCSK Conference, 20th International Federation of Societies of Cosmetic Chemists (IFSCC) Conference, Seoul, Republic of Korea, 1 September 2003; pp. 501–519. [Google Scholar]
- Marquez, G.R.; Di Pierro, P.; Esposito, M.; Mariniello, L.; Porta, R. Application of Transglutaminase-Crosslinked Whey Protein/Pectin Films as Water Barrier Coatings in Fried and Baked Foods. Food Bioprocess. Technol. 2013, 7, 447–455. [Google Scholar] [CrossRef]
- Qu, B.; Zhong, Q. Casein-maltodextrin conjugate as an emulsifier for fabrication of structured calcium carbonate particles as dispersible fat globule mimetics. Food Hydrocoll. 2017, 66, 61–70. [Google Scholar] [CrossRef]
- Carlmark; Malmström, E.E. ATRP Grafting from Cellulose Fibers to Create Block-Copolymer Grafts. Biomacromolecules 2003, 4, 1740–1745. [Google Scholar] [CrossRef]
- Vilaseca, F.; Mendez, J.; Pèlach, A.; Llop, M.; Cañigueral, N.; Gironès, J.; Turon, X.; Mutjé, P. Composite materials derived from biodegradable starch polymer and jute strands. Process Biochem. 2007, 42, 329–334. [Google Scholar] [CrossRef]
- Hu, Y.; Shi, C.-Y.; Xun, X.-M.; Huang, B.-R.; You, S.; Wu, F.-A.; Wang, J. Xylanase-polymer conjugates as new catalysts for xylooligosaccharides production from lignocellulose. Biochem. Eng. J. 2021, 171, 108025. [Google Scholar] [CrossRef]
- Fan, L.; Chen, H.; Wei, S.; Cao, F. Protein-polymer hybrid oil-absorbing gel using hair keratin as macroinitiator by SET-LRP. React. Funct. Polym. 2014, 75, 56–62. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Advantages | Disadvantages |
|---|---|---|
| PEG | High solubility, biocompatibility, and ability to extend protein half-life | Non-biodegradable, polydisperse |
| HPMA | Enhanced drug delivery, versatile applications | Complex synthesis methods |
| Dextran | Biocompatible, biodegradable | Lower mechanical strength |
| PGA | Biodegradable, enzymatically cleavable | Limited scalability |
| Synthesis Technique | Description | Advantages | Limitations | Biomedical Relevance |
|---|---|---|---|---|
| Controlled Radical Polymerization (CRP) | General method using radicals for precise polymer growth | Low polydispersity, tunable properties | Requires specific initiators and control agents | Smart hydrogels, drug delivery |
| ATRP | Uses halogenated initiators with metal catalysts | Precise chain control, low polydispersity | Requires expensive catalysts and controlled conditions | Drug carriers, responsive hydrogels |
| RAFT | Chain transfer agents regulate radical polymerization | High versatility, compatibility with diverse monomers | Chain transfer agent optimization needed | Biodegradable polymers, controlled-release systems |
| Grafting-to | Attaches pre-formed polymers to functional proteins | High yields, straightforward modification | Steric hindrance limits efficiency | PEGylated proteins, enzyme stabilization |
| Grafting-from | Polymerization initiates from protein-linked sites | Site-specific polymer growth, improved purification | Complex initiation control | Amphiphilic protein–polymer micelles |
| Grafting-through | Biomolecules participate as monomers in polymerization | Integrated conjugate networks | Requires biomolecule functionalization | DNA–protein conjugates, nanoparticle systems |
| PEGylation | Attaches PEG to proteins for stability and solubility enhancement | Reduced immunogenicity, extended half-life | Polydispersity, non-biodegradability | Therapeutic protein conjugates, drug delivery |
| Classical PEGylation | Nucleophilic reaction with amino or thiol groups | Simple and efficient | Limited site specificity | PEG-protein therapeutics |
| Bridging PEGylation | Uses disulfide bonds for linking | Maintains protein functionality | Sensitive to reducing conditions | Sustained-release drug carriers |
| Enzymatic PEGylation | Targets glutamine or glycoproteins | High site specificity, homogeneous products | Limited by enzyme availability | Uniform PEGylated proteins |
| Site-Specific Conjugation | Binds polymers to defined protein sites | Enhanced control of bioactivity | Requires functionalized polymers | Targeted drug delivery, responsive hydrogels |
| Cysteine-targeted | Uses thiol-disulfide exchange | Selective conjugation, robust bonds | Limited by cysteine availability | Stabilized protein–polymer conjugates |
| Lysine-targeted | Amide bond formation with amino groups | High reactivity, versatile applications | Potential random conjugation | Long-circulating bioconjugates |
| Material | Function |
|---|---|
| Modified BSA | Protein macroinitiator |
| Cu2+/bipyridine | Catalyst |
| 2-hydroxyethyl methacrylate (HEMA) | Monomer |
| N, N’-methylene diacrylamide | Cross-linker |
| Ascorbic acid | Reducing agent |
| Factor | Description |
|---|---|
| Polymer | Some polymers are cytotoxic due to chemical toxicity or physical interference with cell function. Others are biocompatible and non-toxic. |
| Protein | Some proteins are inherently cytotoxic due to chemical toxicity or immune stimulation. Others are non-toxic. |
| Conjugation method | Conjugation methods that involve chemical crosslinking or toxic reagents can introduce cytotoxic groups into the material. Gentle methods that rely on chemical reactions or physical entrapment can reduce cytotoxicity. |
| Size and shape | Larger conjugates and those with irregular shapes may be more cytotoxic due to their ability to entrap or aggregate cells or disrupt cell function physically. |
| Delivery method | Some delivery methods may be more cytotoxic than others due to the stresses of injection or the presence of foreign substances in the delivery vehicle. |
| Type of Albumin | Polymer | Function |
|---|---|---|
| HAS | poly (β-aminoester Urethane) | Delivery of the Hyperuricemia-diseases treatment uricase (Uox) |
| HAS | (Dex-Mal) polymer | Delivery of the anti-cancer drug DOX |
| HAS | DEX (VS) + PEG | Delivery of the anti-cancer drug DOX |
| HAS | 4-arm PEG-maleimide | Delivery of the anti-cancer protein TRAIL |
| BSA | Triblock copolymer (PCLA) | The encapsulation and delivery of pDNA vaccines |
| BSA | DMDOMA polymer | The delivery of CL075 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alayoubi, O.; Poyraz, Y.; Hassan, G.; Gül, S.B.; Çalhan, N.; Mert Şahin, N.M.; Gautam, M.; Kutlu, A.; Özuğur Uysal, B.; Akten, E.D.; et al. Hydrogels from Protein–Polymer Conjugates: A Pathway to Next-Generation Biomaterials. Gels 2025, 11, 96. https://doi.org/10.3390/gels11020096
Alayoubi O, Poyraz Y, Hassan G, Gül SB, Çalhan N, Mert Şahin NM, Gautam M, Kutlu A, Özuğur Uysal B, Akten ED, et al. Hydrogels from Protein–Polymer Conjugates: A Pathway to Next-Generation Biomaterials. Gels. 2025; 11(2):96. https://doi.org/10.3390/gels11020096
Chicago/Turabian StyleAlayoubi, Oubadah, Yağmur Poyraz, Gana Hassan, Sümeyye Berfin Gül, Nergiz Çalhan, Naz Mina Mert Şahin, Megha Gautam, Aylin Kutlu, Bengü Özuğur Uysal, Ebru Demet Akten, and et al. 2025. "Hydrogels from Protein–Polymer Conjugates: A Pathway to Next-Generation Biomaterials" Gels 11, no. 2: 96. https://doi.org/10.3390/gels11020096
APA StyleAlayoubi, O., Poyraz, Y., Hassan, G., Gül, S. B., Çalhan, N., Mert Şahin, N. M., Gautam, M., Kutlu, A., Özuğur Uysal, B., Akten, E. D., & Pekcan, Ö. (2025). Hydrogels from Protein–Polymer Conjugates: A Pathway to Next-Generation Biomaterials. Gels, 11(2), 96. https://doi.org/10.3390/gels11020096