Hyaluronic Acid-Poly(N-acryloyl glycinamide) Copolymers as Sources of Degradable Thermoresponsive Hydrogels for Therapy



Abstract
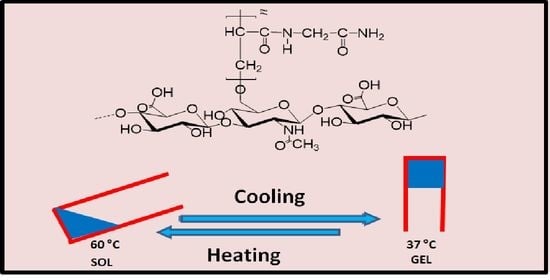
:
1. Introduction
2. Results and Discussion
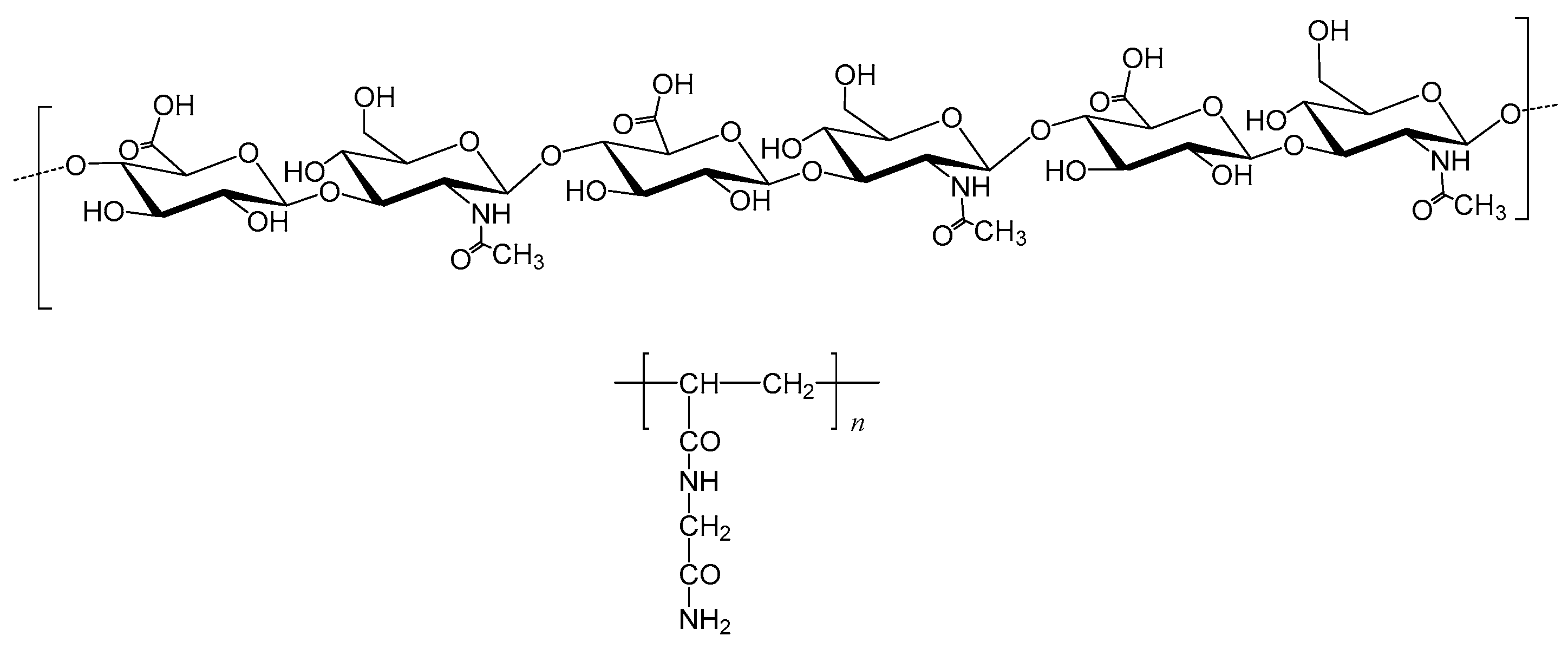
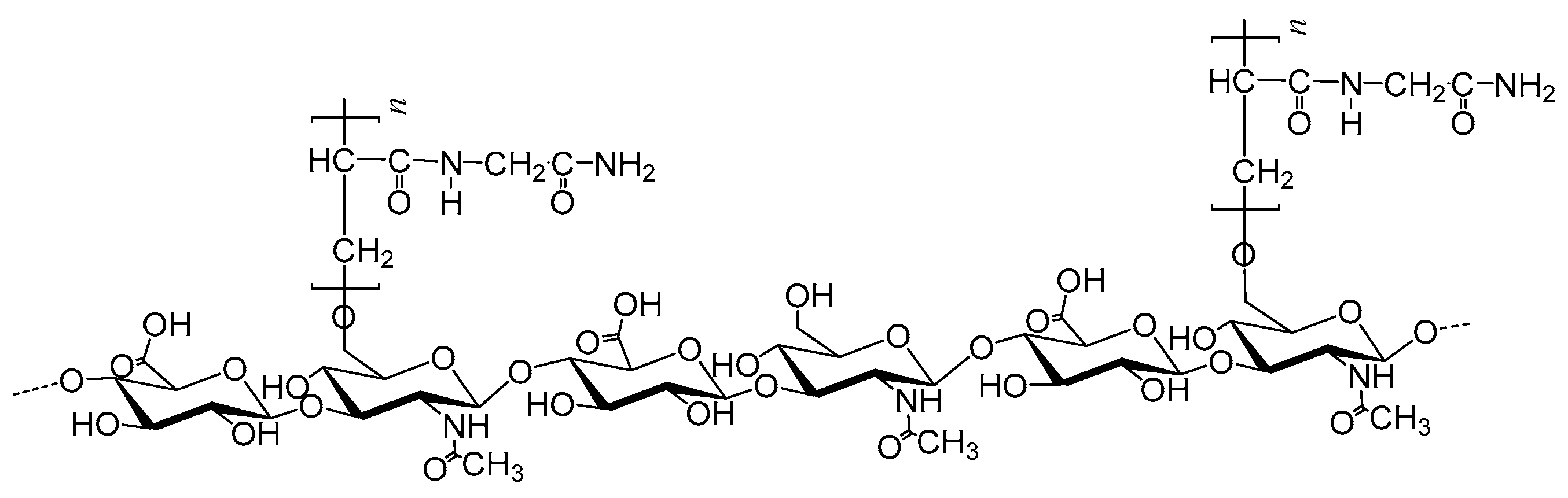
2.1. Synthesis
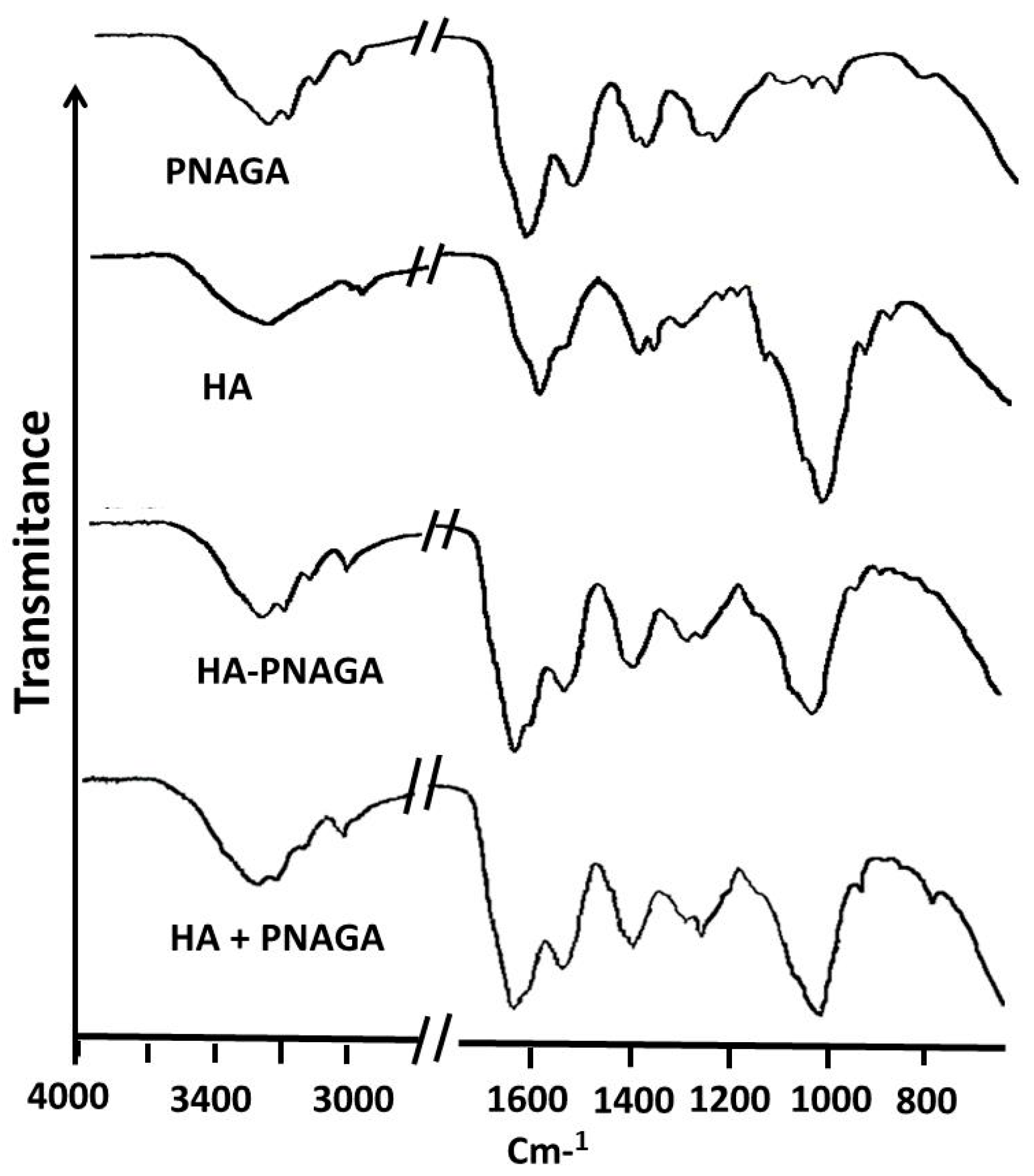
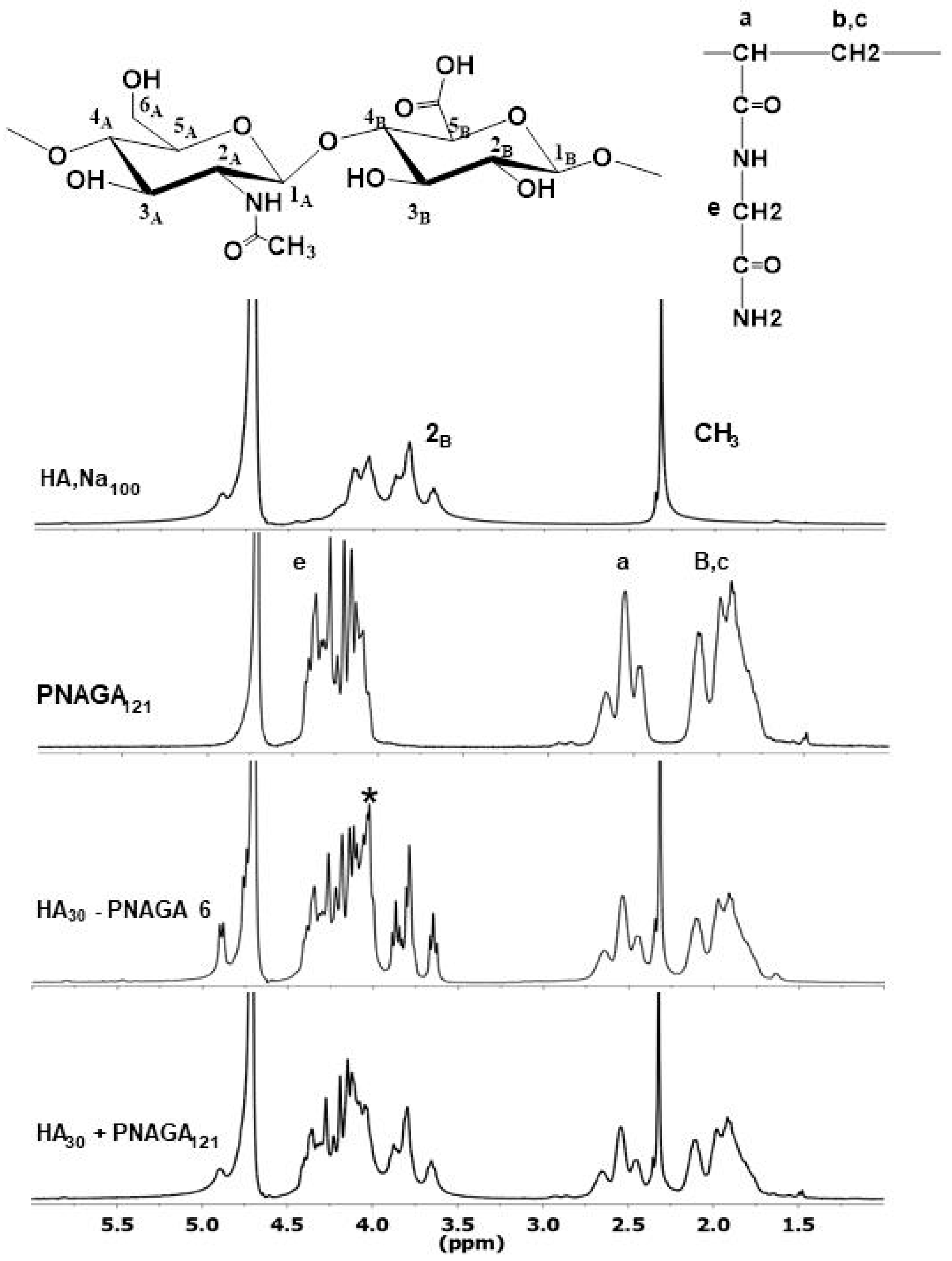
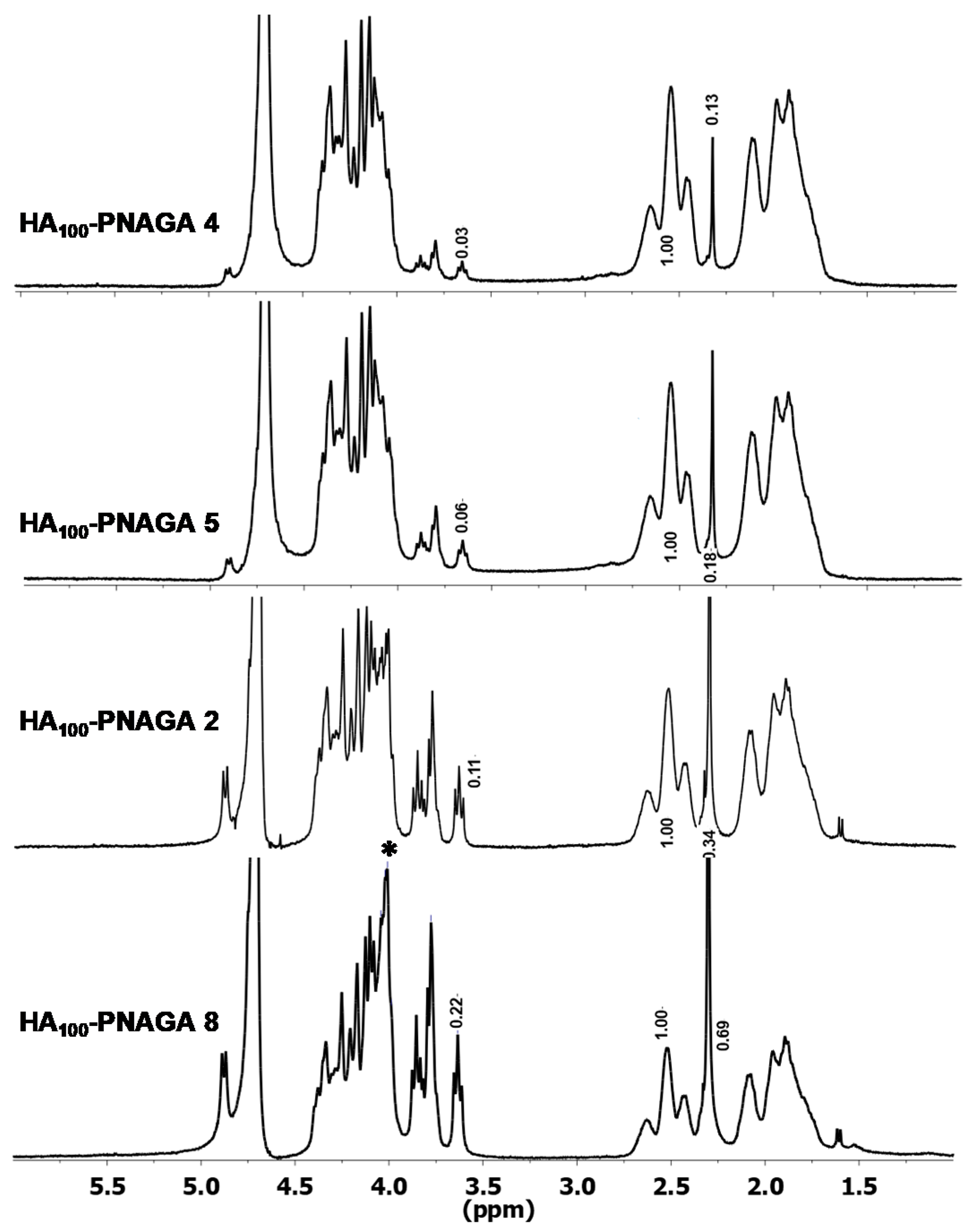
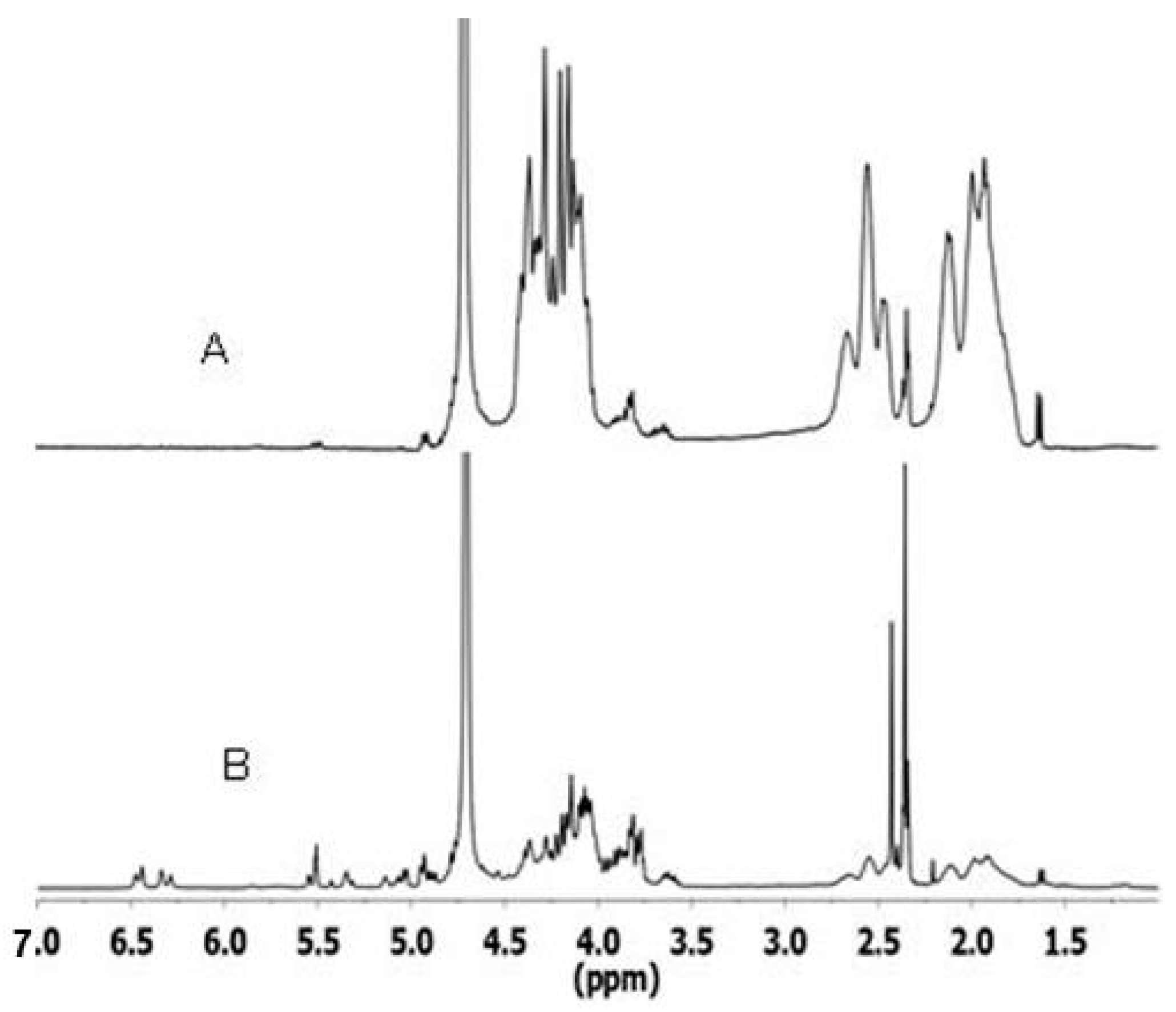
2.2. Spectroscopic Analyses
2.3. Indirect Demonstration of the Grafting of PNAGA onto HA Macromolecules
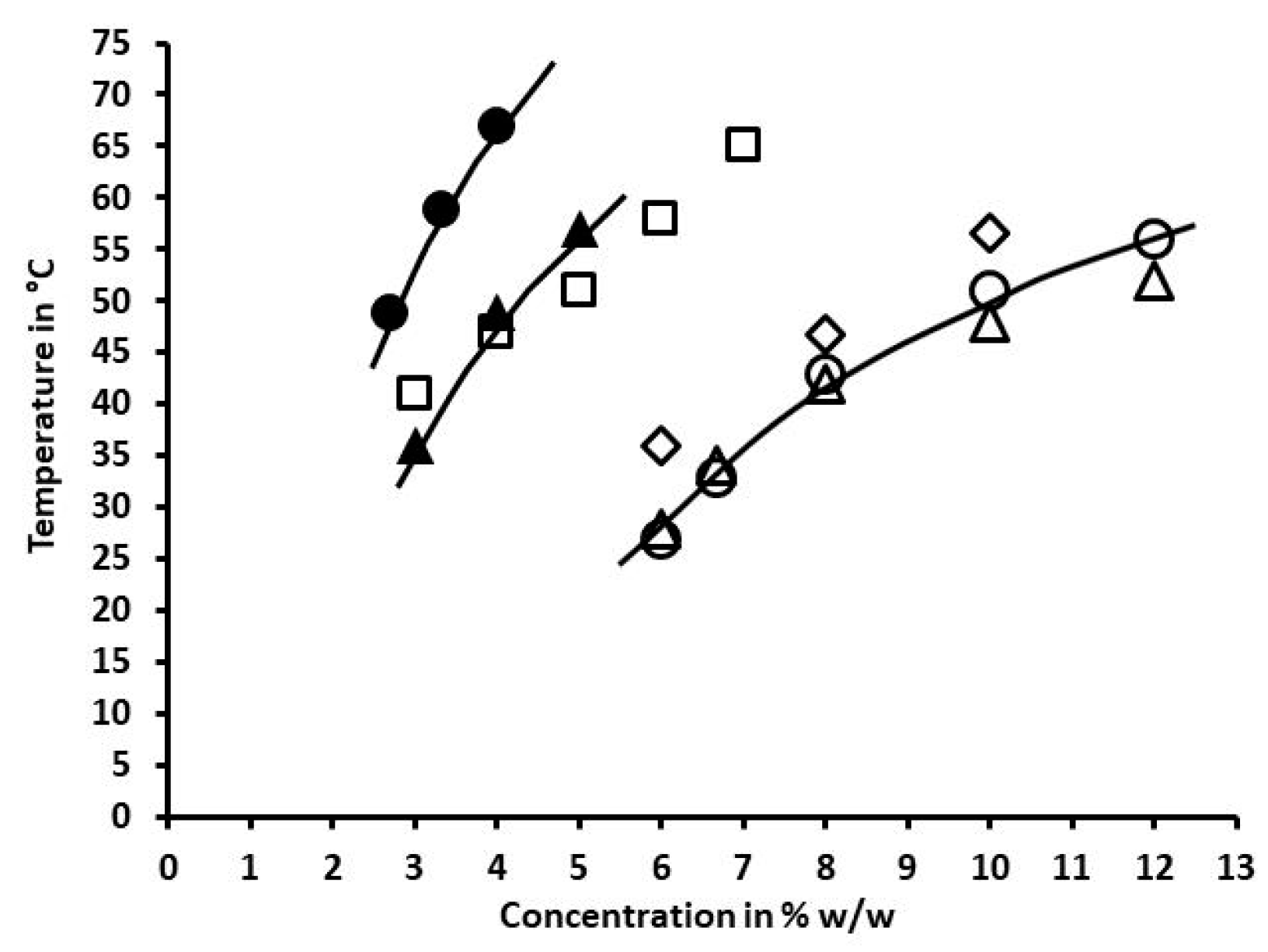
2.3.1. Physical Aspect
2.3.2. Molar Mass Determination
2.3.3. Ionic Interaction with Cationic Poly(l-Lysine)
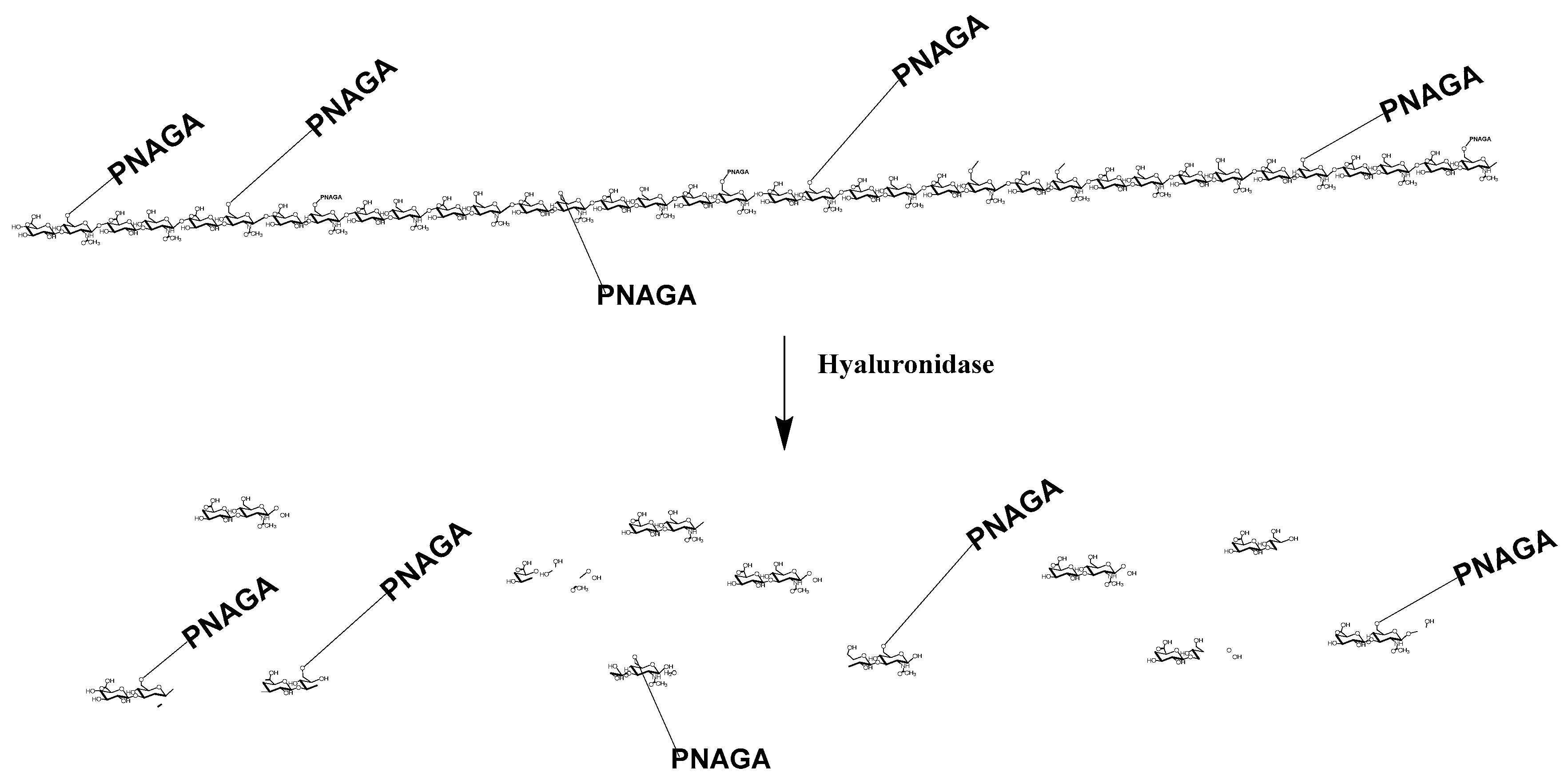
2.3.4. Degradation by Hyaluronidase
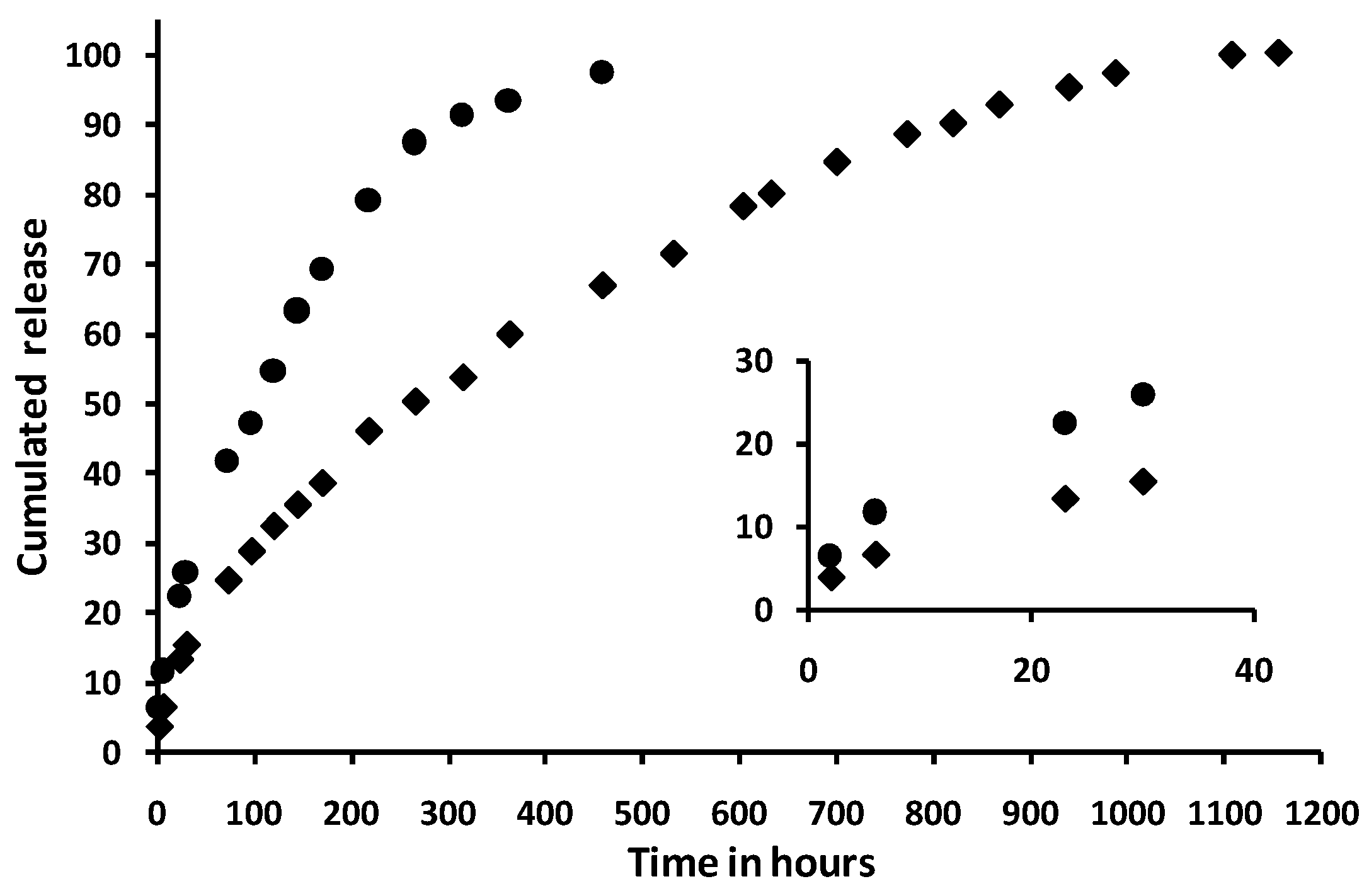
2.4. Ability to Sustaining the Release of a Drug
3. Conclusions
4. Materials and Methods
4.1. Materials
4.1.1. Chemicals
4.1.2. Synthesis of HA-PNAGA Copolymer
4.2. Methods
4.2.1. Spectral Characterizations
4.2.2. Thermal Characterizations
4.2.3. Size Exclusion Chromatography (SEC)
4.2.4. Enzymatic Degradation
4.2.5. Formulation of Prednisolone Hydrogels
4.2.6. Prednisolone Sustained Release Profiles
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; The “Gold Book”; Blackwell Scientific Publications: Oxford, UK, 1997; ISBN 0-9678550-9-8. [Google Scholar] [CrossRef]
- Laftah, W.A.; Hashim, S.; Ibrahim, A.N. Polymer Hydrogels: A Review. Polym. Plast. Technol. Eng. 2011, 50, 1475–1486. [Google Scholar]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23. [Google Scholar]
- Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them. Gels 2017, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Vashist, A.; Vashist, A.; Gupta, Y.K.; Amad, S. Recent advances in hydrogel based drug delivery systems for the human body. J. Mater. Chem. B 2014, 2, 147–166. [Google Scholar] [PubMed]
- Amin, S.; Rajabnezhad, S.; Kohli, K. Hydrogels as potential drug delivery systems. Sci. Res. Essay 2009, 3, 1175–1183. [Google Scholar]
- Zavan, B.; Cortivo, R.; Abatangelo, G. Hydrogels and Tissue Engineering. In Hydrogels; Springer: Milano, Italy, 2009; pp. 1–8. ISBN 978-88-470-1103-8. [Google Scholar] [CrossRef]
- Sood, N.; Bhardwaj, A.; Mehta, S.; Mehta, A. Stimuli-responsive hydrogels in drug delivery and tissue engineering. Drug Deliv. 2016, 23, 748–770. [Google Scholar] [CrossRef] [Green Version]
- Vashist, A.; Kaushik, A.; Jayant, R.D.; Vashist, A.; Ghosal, A.; Nair, M. Hydrogels: Stimuli Responsive to on-Demand Drug Delivery Systems. In Advances in Personalized Nanotherapeutics; Kaushik, A., Jayant, R., Nair, M., Eds.; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Taylor, M.J.; Tomlins, P.; Sahota, T.S. Thermoresponsive gels. Gels 2017, 3, 4. [Google Scholar] [CrossRef]
- Aseyev, V.; Tenhu, H.; Winnik, F.M. Non-ionic Thermoresponsive Polymers in Water. Adv. Polym. Sci. 2010, 242, 29–89. [Google Scholar]
- Shimada, N.; Kidoaki, S.; Maruyama, A. Smart hydrogels exhibiting UCST-type volume changes under physiologically relevant conditions. RSC Adv. 2014, 4, 52346–52348. [Google Scholar]
- Miyawaki, O.; Omote, C.; Matsuhira, K. Thermodynamic analysis of sol-gel transition of gelatin in terms of water activity in various solutions. Biopolymers 2015, 103, 685–691. [Google Scholar] [CrossRef]
- Boustta, M.; Colombo, P.E.; Lenglet, S.; Poujol, S.; Vert, M. Versatile UCST-based thermoresponsive hydrogels for loco-regional sustained drug delivery. J. Control. Rel. 2014, 174, 1–6. [Google Scholar]
- Seuring, J.; Bayer, F.M.; Huber, K.; Agarwal, S. Upper Critical Solution Temperature of Poly(N-acryloyl glycinamide) in Water: A Concealed Property. Macromolecules 2012, 45, 374–384. [Google Scholar] [CrossRef]
- Vert, M.; Doi, Y.; Hellwich, K.H.; Hess, M.; Hodge, P.; Kubisa, P.; Rinaudo, M.; Schue, F. Terminology for biorelated polymers and applications (IUPAC Recommendations 2012). Pure Appl. Chem. 2012, 84, 377–408. [Google Scholar]
- Payan, E.; Jouzeau, J.Y.; Lapicque, F.; Muller, N.; Netter, P. Hyaluronidase degradation of hyaluronic acid from different sources: Influence of the hydrolysis conditions on the production and the relative proportions of tetra- and hexasaccharide produced. Intern. J. Biochem. 1993, 25, 325–329. [Google Scholar]
- Awwad, S.; Abdullah, A.; Angkawinitwong, U.; Ukrit, K.; Khaw, P.T.; Brocchini, S. In situ antibody-loaded hydrogel for intravitreal delivery. Eur. J. Pharm. Sci. 2019, 137, 0928–0987. [Google Scholar]
- Tiwari, S.; Bahadur, P. Modified hyaluronic acid based materials for biomedical applications. Intern. J. Biol. Macromol. 2009, 121, 556–571. [Google Scholar]
- Huaping, T.; Ramireza, C.M.; Natasa, M.; HanLia, L.J.; Rubinab, J.P.; Kacey, G.M. Thermosensitive injectable hyaluronic acid hydrogel for adipose tissue engineering. Biomaterials 2009, 30, 6844–6853. [Google Scholar]
- Lü, S.; Li, B.; Ni, B.; Sun, Z.; Liu, M.; Wang, Q. Thermoresponsive injectable hydrogel for three-dimensional cell culture: Chondroitin sulfate bioconjugated with poly(N-isopropylacrylamide) synthesized by RAFT polymerization. Soft Matter 2011, 7, 10763–10772. [Google Scholar]
- Haas, H.C.; Chiklis, C.K.; Moreau, R.D. Synthetic thermally reversible gel systems. III. J. Polym. Sci. Part A-1 Polym. Chem. 1970, 8, 1131–1145. [Google Scholar]
- Colombani, D. Chain-growth control in free radical polymerization. Progr. Polym. Sci. 1997, 22, 1649–1720. [Google Scholar] [CrossRef]
- Glatzel, S.; Laschewsky, A.; Lutz, J.-F. Well-defined uncharged polymers with a sharp UCST in water and in physiological milieu. Macromolecules 2011, 44, 413–415. [Google Scholar]
- Leclercq, L.; Boustta, M.; Vert, M. Dynamics of polyelectrolyte complex formation and stability when a polycation is progressively added to a polyanion under physico-chemical conditions modeling blood. J. Bioact. Compat. Polym. 2011, 26, 3–19. [Google Scholar]
- Vert, M. Polymeric biomaterials: Strategies of the past vs. strategies of the future. Prog. Polym. Sci. 2007, 32, 755–767. [Google Scholar]
- Vert, M. Degradable and bioresorbable polymers in surgery and in pharmacology: Beliefs and facts. J. Mater. Sci. Mater. Med. 2009, 20, 437–446. [Google Scholar] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymers | HA,Na a g | NAGA a g | HA/ HA + NAGA b W/W % | HA/ HA + NAGA c g W/W % (Moldisac%) | Yield % ± 2 | Gel → Sol Transition Temperature d °C |
|---|---|---|---|---|---|---|
| HA30 − PNAGA 1 | 0.3 | 1.2 | 20 | 97 | 55–56 | |
| HA100 − PNAGA 2 | 0.375 | 1.5 | 20 | 23 (7.4) | 96 | 71–72 |
| HA100 − PNAGA 3 | 0.065 | 2.0 | 3.15 | 96 | 66–68 | |
| HA100 − PNAGA 4 | 0.25 | 2.5 | 9.18 | 9.2 (2.93) | 95 | 83–84 |
| HA100 − PNAGA 5 | 0.4 | 2.5 | 13.8 | 17.7 (5.7) | 95 | 85–87 |
| HA30 − PNAGA 6 | 0.4 | 0.4 | 50 | 54.4 (17.4) | 97 | 43–45 e |
| HA100 − PNAGA 7 | 0.4 | 0.4 | 50 | 98 | 41–42 e | |
| HA100 − PNAGA 8 | 0.4 | 0.4 | 50 | 56.4 (18) | 94 | 42–43 e |
| HA2000 − PNAGA 9 | 0.4 | 2.5 | 13.8 | 96 | 77–79 | |
| HA2000 − PNAGA 10 | 0.2 | 2.5 | 9.18 | 9.2 (2.9) | 95 | 73–75 |
| HA30 + PNAGA121 | 0.01 | 0.01 | 50 | 50 (16 ± 1) | - | - |
| HA100 + PNAGA121 | 0.01 | 0.01 | 50 | 53 (17 ± 1) | - | 58–59 f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boustta, M.; Vert, M. Hyaluronic Acid-Poly(N-acryloyl glycinamide) Copolymers as Sources of Degradable Thermoresponsive Hydrogels for Therapy. Gels 2020, 6, 42. https://doi.org/10.3390/gels6040042
Boustta M, Vert M. Hyaluronic Acid-Poly(N-acryloyl glycinamide) Copolymers as Sources of Degradable Thermoresponsive Hydrogels for Therapy. Gels. 2020; 6(4):42. https://doi.org/10.3390/gels6040042
Chicago/Turabian StyleBoustta, Mahfoud, and Michel Vert. 2020. "Hyaluronic Acid-Poly(N-acryloyl glycinamide) Copolymers as Sources of Degradable Thermoresponsive Hydrogels for Therapy" Gels 6, no. 4: 42. https://doi.org/10.3390/gels6040042
APA StyleBoustta, M., & Vert, M. (2020). Hyaluronic Acid-Poly(N-acryloyl glycinamide) Copolymers as Sources of Degradable Thermoresponsive Hydrogels for Therapy. Gels, 6(4), 42. https://doi.org/10.3390/gels6040042