

Novel Silica Hybrid Xerogels Prepared by Co-Condensation of TEOS and ClPhTEOS: A Chemical and Morphological Study

 ,
,  ,
,  and
and 
Abstract
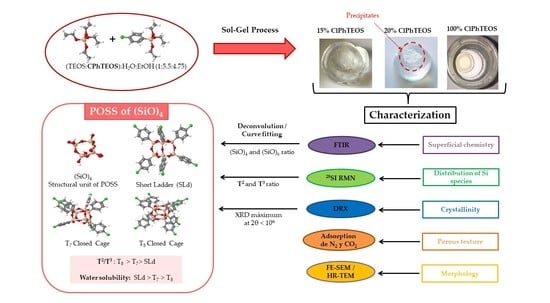
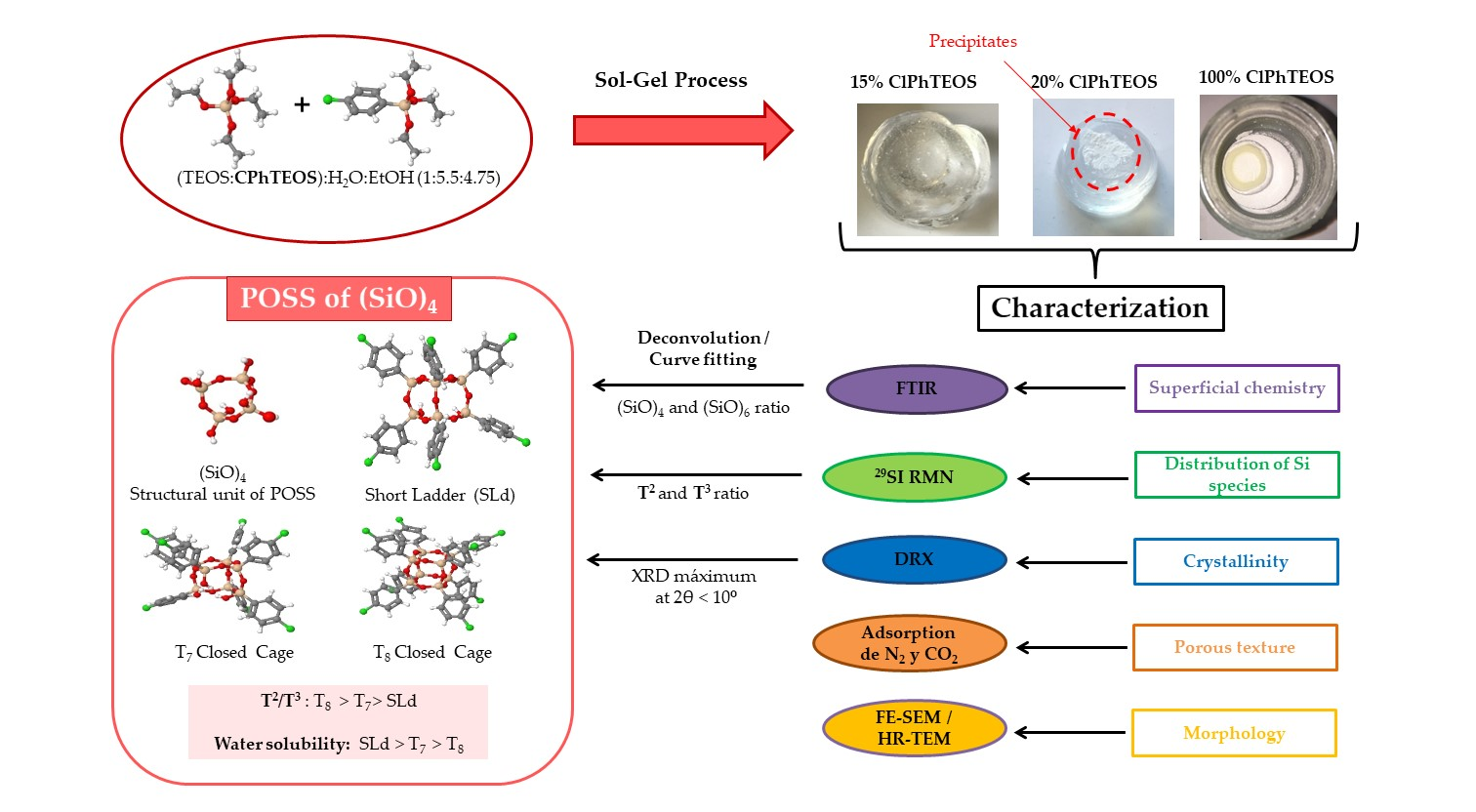
1. Introduction
2. Results and Discussion
2.1. FT-IR
2.2. 29Si Nuclear Magnetic Resonance (NMR)
2.3. X-ray Diffraction (XRD)
2.4. Helium Pycnometry
2.5. N2 and CO2 Adsorption Isotherms
2.6. Microscopy
2.6.1. Field-Emission Scanning Electron Microscopy (FE-SEM)
2.6.2. High Resolution-Transmission Electron Microscopy (HR-TEM)
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Silicon Hybrid Xerogels
4.3. Characterization of Silicon Hybrid Xerogels
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hannachi, Y.; Hafidh, A.; Ayed, S. Effectiveness of novel xerogels adsorbents for cadmium uptake from aqueous solution in batch and column modes: Synthesis, characterization, equilibrium, and mechanism analysis. Chem. Eng. Res. Des. 2019, 143, 11–23. [Google Scholar] [CrossRef]
- Kobylinska, N.G.; Kessler, V.G.; Seisenbaeva, G.A.; Dudarko, O.A. In Situ Functionalized Mesoporous Silicas for Sustainable Remediation Strategies in Removal of Inorganic Pollutants from Contaminated Environmental Water. ACS Omega 2022, 7, 23576–23590. [Google Scholar] [CrossRef]
- Wojciechowska, P.; Cierpiszewski, R. Gelatin—Siloxane Hybrid Monoliths as Novel Heavy Metal Adsorbents. Appl. Sci. 2022, 12, 1258. [Google Scholar] [CrossRef]
- Didó, C.A.; Coelho, F.L.; Closs, M.B.; Deon, M.; Horowitz, F.; Bernardi, F.; Schneider, P.H.; Benvenutti, E. V Strategy to isolate ionic gold sites on silica surface: Increasing their efficiency as catalyst for the formation of 1,3-diynes. Appl. Catal. A Gen. 2020, 594, 117444. [Google Scholar] [CrossRef]
- Estevez, R.; Luna, D.; Bautista, F.M. Sulfonated organosilica-aluminum phosphates as useful catalysts for acid-catalyzed reactions: Insights into the effect of synthesis parameters on the final catalyst. Catal. Today 2022, 390–391, 12–21. [Google Scholar] [CrossRef]
- Liu, M.; Wang, L.; Zhang, L.; Zhao, Y.; Chen, K.; Li, Y. In-Situ Silica Xerogel Assisted Facile Synthesis of Fe-N-C Catalysts with Dense Fe-Nx Active Sites for Efficient Oxygen Reduction. Small 2022, 18, 202104934. [Google Scholar] [CrossRef]
- Song, L.; Jia, Y.; Jin, X.; Wang, X.; Chen, M.; Wu, Y. Novel hybrid xerogel with CaF2:Tb3+ nanoparticles covalently embedded into silicon-oxygen network through sol-gel process. J. Lumin. 2022, 250, 119071. [Google Scholar] [CrossRef]
- Vilela, R.R.C.; Zanoni, K.P.S.; de Oliveira, M.; de Vicente, F.S.; de Camargo, A.S.S. Structural and photophysical characterization of highly luminescent organosilicate xerogel doped with Ir(III) complex. J. Sol-Gel Sci. Technol. 2022, 102, 236–248. [Google Scholar] [CrossRef]
- Santos, J.F.M.; Zanuto, V.S.; Ventura, M.; Bramorski, C.B.; Catunda, T. Quantum yield measurements by thermal lens in highly absorbing samples: The case of highly doped rhodamine B organic/silica xerogels. Phys. Rev. Mater. 2019, 3, 115201. [Google Scholar] [CrossRef]
- Alcantara-garcia, A.; Garcia-casas, A.; Jimenez-morales, A. The effect of the organosilane content on the barrier features of sol-gel anticorrosive coatings applied on carbon steel. Prog. Org. Coat. 2020, 139, 105418. [Google Scholar] [CrossRef]
- Scotland, K.M.; Shetranjiwalla, S.; Vreugdenhil, A.J. Curable hybrid materials for corrosion protection of steel: Development and application of UV-cured 3-methacryloxypropyltrimethoxysilane-derived coating. J. Coat. Technol. Res. 2020, 17, 977–989. [Google Scholar] [CrossRef]
- Rodič, P.; Milošev, I.; Lekka, M.; Andreatta, F.; Fedrizzi, L. Corrosion behaviour and chemical stability of transparent hybrid sol-gel coatings deposited on aluminium in acidic and alkaline solutions. Prog. Org. Coat. 2018, 124, 286–295. [Google Scholar] [CrossRef]
- Hao, S.; Hu, C.; Lin, X. Resistance to Growth of Molds for Wood Modified with Hydrophobic Hybrid Silica Gel Containing Copper Amine Complexes. Materials 2021, 14, 577. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yue, W.; Zhang, S.; Zhang, Y.; Li, C.; Su, W. Modification of Silica Xerogels with Polydopamine for Lipase B from Candida antarctica Immobilization. Catalysts 2021, 11, 1463. [Google Scholar] [CrossRef]
- Hatano, K.; Teraki, M.; Nakajima, D.; Yamatsu, T. Controlled release of molasses melanoidin-like product from hybrid organic–inorganic silica xerogels and its application to the phytoextraction of lead through the Indian mustard. Environ. Sci. Pollut. Res. 2021, 28, 37562–37569. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Moreno, A.; Reyes-Peces, M.V.; Vilches-Pérez, J.I.; Fernández-Montesinos, R.; Pinaglia-Tobaruela, G.; Salido, M.; de la Rosa-Fox, N.; Piñero, M. Effect of Washing Treatment on the Textural Properties and Bioactivity of Silica/Chitosan/TCP Xerogels for Bone Regeneration. Int. J. Mol. Sci. 2021, 22, 8321. [Google Scholar] [CrossRef] [PubMed]
- Kapusuz, D. Sol-gel derived silica/polyethylene glycol hybrids as potential oligonucleotide vectors. J. Mater. Res. 2019, 34, 3787–3797. [Google Scholar] [CrossRef]
- Goel, H.; Santhiya, D. Effect of pH on bio-inspired synthesis of L-Lysine templated bioactive glass hybrid xerogels for tailored textural and rheological properties. Mater. Chem. Phys. 2022, 281, 125828. [Google Scholar] [CrossRef]
- Wendels, S.; de Souza Porto, D.; Avérous, L. Synthesis of biobased and hybrid polyurethane xerogels from bacterial polyester for potential biomedical applications. Polymers 2021, 13, 4256. [Google Scholar] [CrossRef]
- Judeinstein, P.; Sanchez, C. Hybrid organic-inorganic materials: A land of multidisciplinarity. J. Mater. Chem. 1996, 6, 511–525. [Google Scholar] [CrossRef]
- Kaya, G.G.; Deveci, H. Synergistic effects of silica aerogels/xerogels on properties of polymer composites: A review. J. Ind. Eng. Chem. 2020, 89, 13–27. [Google Scholar] [CrossRef]
- Kolle, J.M.; Sayari, A. Dry gel grafting of mesoporous silica: Application to amine-based CO2 adsorbents. Microporous Mesoporous Mater. 2022, 343, 112195. [Google Scholar] [CrossRef]
- Jahandar, M.; Ziaei-azad, H.; Sayari, A. Unprecedented improvement of the hydrothermal stability of amine-grafted MCM-41 silica for CO2 capture via aluminum incorporation. Chem. Eng. J. 2022, 450, 138393. [Google Scholar] [CrossRef]
- Yan, J.; Kuang, M.; Zhu, Y.; Chen, Y.; Zhang, C.; Ma, H.; Zhang, X.; Kong, L. Facile, tunable, and environmental friendly synthesis of silica/resorcinol–formaldehyde hybrid xerogels with ultra-low shrinkage using a cationic polyelectrolyte as a soft template. J. Porous Mater. 2022. [Google Scholar] [CrossRef]
- Flores-López, S.L.; Villanueva, S.F.; Rey-Raap, N.; Arenillas, A. Hybrid rf-si xerogels: A cost-effective proposal for insulator materials. Materials 2022, 15, 265. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, M.; Vaezi, M.R.; Kazemzadeh, A. The influence of the hydrophobic agent, catalyst, solvent and water content on the wetting properties of the silica films prepared by one-step sol-gel method. Appl. Surf. Sci. 2015, 326, 99–106. [Google Scholar] [CrossRef]
- Dudás, Z.; Len, A.; Ianăși, C.; Paladini, G. Structural modifications caused by the increasing MTES amount in hybrid MTES/TEOS-based silica xerogels. Mater. Charact. 2020, 167, 33–36. [Google Scholar] [CrossRef]
- Mageste, R.L.; Sampaio, D.; Mayrink, T.F.; Ahmed, H.; Palhares, H.G.; Nunes, E.H.M.; Houmard, M. Influence of the alcoholic solvent and gelation temperature on the structural and water vapor adsorption properties of silica adsorbents synthesized by sol-gel method without catalyst. Microporous Mesoporous Mater. 2022, 342, 112114. [Google Scholar] [CrossRef]
- Innocenzi, P. The Sol-Gel Transition, 2nd ed.; Springer: Sassari, Italy, 2019; ISBN 978-3-030-20029-9. [Google Scholar]
- Zhao, X.; Wang, Y.; Luo, J.; Wang, P.; Xiao, P.; Jiang, B. The Influence of Water Content on the Growth of the Hybrid-Silica Particles by Sol-Gel Method. Silicon 2020, 13, 3413–3421. [Google Scholar] [CrossRef]
- Estella, J.; Echeverría, J.C.; Laguna, M.; Garrido, J.J. Silica xerogels of tailored porosity as support matrix for optical chemical sensors. Simultaneous effect of pH, ethanol:TEOS and water:TEOS molar ratios, and synthesis temperature on gelation time, and textural and structural properties. J. Non-Cryst. Solids 2007, 353, 286–294. [Google Scholar] [CrossRef]
- Musgo, J.; Echeverría, J.C.; Estella, J.; Laguna, M.; Garrido, J.J. Ammonia-catalyzed silica xerogels: Simultaneous effects of pH, synthesis temperature, and ethanol:TEOS and water:TEOS molar ratios on textural and structural properties. Microporous Mesoporous Mater. 2009, 118, 280–287. [Google Scholar] [CrossRef]
- Echeverría, J.C.; Estella, J.; Barbería, V.; Musgo, J.; Garrido, J.J. Synthesis and characterization of ultramicroporous silica xerogels. J. Non-Cryst. Solids 2010, 356, 378–382. [Google Scholar] [CrossRef]
- Echeverría, J.C.; Moriones, P.; Arzamendi, G.; Garrido, J.J.; Gil, M.J.; Cornejo, A.; Martínez-Merino, V. Kinetics of the acid-catalyzed hydrolysis of tetraethoxysilane (TEOS) by 29Si NMR spectroscopy and mathematical modeling. J. Sol-Gel Sci. Technol. 2018, 86, 316–328. [Google Scholar] [CrossRef]
- Rios, X.; Moriones, P.; Echeverría, J.C.; Luquín, A.; Laguna, M.; Garrido, J.J. Characterisation of hybrid xerogels synthesised in acid media using methyltriethoxysilane (MTEOS) and tetraethoxysilane (TEOS) as precursors. Adsorption 2011, 17, 583–593. [Google Scholar] [CrossRef]
- Moriones, P.; Echeverria, J.C.; Garrido, J.B.P.J.J. Phenyl siloxane hybrid xerogels: Structure and porous texture. Adsorption 2020, 26, 177–188. [Google Scholar] [CrossRef]
- Cruz-Quesada, G.; Espinal-Viguri, M.; López-Ramón, M.; Garrido, J. Novel Organochlorinated Xerogels: From Microporous Materials to Ordered Domains. Polymers 2021, 13, 1415. [Google Scholar] [CrossRef]
- Rios, X.; Moriones, P.; Echeverría, J.C.; Luquin, A.; Laguna, M.; Garrido, J.J. Ethyl group as matrix modifier and inducer of ordered domains in hybrid xerogels synthesised in acidic media using ethyltriethoxysilane (ETEOS) and tetraethoxysilane (TEOS) as precursors. Mater. Chem. Phys. 2013, 141, 166–174. [Google Scholar] [CrossRef]
- Ospino, I.; Luquin, A.; Jiménez-Ruiz, M.; Pérez-Landazábal, J.I.; Recarte, V.; Echeverría, J.C.; Laguna, M.; Urtasun, A.A.; Garrido, J.J. Computational Modeling and Inelastic Neutron Scattering Contributions to the Study of Methyl-silica Xerogels: A Combined Theoretical and Experimental Analysis. J. Phys. Chem. C 2017, 121, 22836–22845. [Google Scholar] [CrossRef]
- Cruz-Quesada, G.; Espinal-Viguri, M.; López-Ramón, M.; Garrido, J. Hybrid Xerogels: Study of the Sol-Gel Process and Local Structure by Vibrational Spectroscopy. Polymers 2021, 13, 2082. [Google Scholar] [CrossRef]
- Estella, J.; De Vicente, P.; Echeverría, J.C.; Garrido, J.J. A fibre-optic humidity sensor based on a porous silica xerogel film as the sensing element. Sens. Actuators B Chem. 2010, 149, 122–128. [Google Scholar] [CrossRef]
- Echeverría, J.C.; Faustini, M.; Garrido, J.J. Effects of the porous texture and surface chemistry of silica xerogels on the sensitivity of fiber-optic sensors toward VOCs. Sens. Actuators B Chem. 2016, 222, 1166–1174. [Google Scholar] [CrossRef]
- Echeverría, J.C.; Calleja, I.; Moriones, P.; Garrido, J.J. Fiber optic sensors based on hybrid phenyl-silica xerogel films to detect n-hexane: Determination of the isosteric enthalpy of adsorption. Beilstein J. Nanotechnol. 2017, 8, 475–484. [Google Scholar] [CrossRef]
- Fidalgo, A.; Ciriminna, R.; Ilharco, L.M.; Pagliaro, M. Role of the alkyl-alkoxide precursor on the structure and catalytic properties of hybrid sol-gel catalysts. Chem. Mater. 2005, 17, 6686–6694. [Google Scholar] [CrossRef]
- Fidalgo, A.; Ilharco, L.M. Chemical Tailoring of Porous Silica Xerogels: Local Structure by Vibrational Spectroscopy. Chem. A Eur. J. 2004, 10, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Innocenzi, P. Infrared spectroscopy of sol–gel derived silica-based films: A spectra-microstructure overview. J. Non-Cryst. Solids 2003, 316, 309–319. [Google Scholar] [CrossRef]
- Lin, W.; Zheng, J.; Zhuo, J.; Chen, H.; Zhang, X. Characterization of sol-gel ORMOSIL antireflective coatings from phenyltriethoxysilane and tetraethoxysilane: Microstructure control and application. Surf. Coat. Technol. 2018, 345, 177–182. [Google Scholar] [CrossRef]
- Launer, P.J.; Arkles, B. Infrared Analysis of Organosilicon Compounds. In Silicon Compd. Silanes Silicones, 3rd ed.; Arkles, B., Larson, G.L., Eds.; Gelest, Inc.: Morrisville, PA; USA, 2013; pp. 175–178. ISBN 978-0-578-12235-9. [Google Scholar]
- Park, E.S.; Ro, H.W.; Nguyen, C.V.; Jaffe, R.L.; Yoon, D.Y. Infrared spectroscopy study of microstructures of poly(silsesquioxane)s. Chem. Mater. 2008, 20, 1548–1554. [Google Scholar] [CrossRef]
- Guo, M.; Kanezashi, M.; Nagasawa, H.; Yu, L.; Yamamoto, K.; Gunji, T.; Ohshita, J.; Tsuru, T. Tailoring the microstructure and permeation properties of bridged organosilica membranes via control of the bond angles. J. Memb. Sci. 2019, 584, 56–65. [Google Scholar] [CrossRef]
- Hayami, R.; Ideno, Y.; Sato, Y.; Tsukagoshi, H.; Yamamoto, K.; Gunji, T. Soluble ethane-bridged silsesquioxane polymer by hydrolysis–condensation of bis(trimethoxysilyl)ethane: Characterization and mixing in organic polymers. J. Polym. Res. 2020, 27, 316. [Google Scholar] [CrossRef]
- Marchesi, S.; Carniato, F.; Palin, L.; Boccaleri, E. POSS as building-blocks for the preparation of polysilsesquioxanes through an innovative synthetic approach. Dalt. Trans. 2015, 44, 2042–2046. [Google Scholar] [CrossRef]
- Choi, S.S.; Lee, A.S.; Lee, H.S.; Baek, K.Y.; Choi, D.H.; Hwang, S.S. Synthesis and characterization of ladder-like structured polysilsesquioxane with carbazole group. Macromol. Res. 2011, 19, 261–265. [Google Scholar] [CrossRef]
- Wu, J.; Mather, P.T. POSS polymers: Physical properties and biomaterials applications. Polym. Rev. 2009, 49, 25–63. [Google Scholar] [CrossRef]
- Choi, S.S.; Lee, A.S.; Hwang, S.S.; Baek, K.Y. Structural Control of Fully Condensed Polysilsesquioxanes: Ladder-like vs Cage Structured Polyphenylsilsesquioxanes. Macromolecules 2015, 48, 6063–6070. [Google Scholar] [CrossRef]
- Jung, J.; Won, J.; Hwang, S.S. Highly selective composite membranes using ladder-like structured polysilsesquioxane for a non-aqueous redox flow battery. J. Memb. Sci. 2020, 595, 117520. [Google Scholar] [CrossRef]
- Depla, A.; Verheyen, E.; Veyfeyken, A.; Van Houteghem, M.; Houthoofd, K.; Van Speybroeck, V.; Waroquier, M.; Kirschhock, C.E.A.; Martens, J.A. UV-Raman and 29Si NMR spectroscopy investigation of the nature of silicate oligomers formed by acid catalyzed hydrolysis and polycondensation of tetramethylorthosilicate. J. Phys. Chem. C 2011, 115, 11077–11088. [Google Scholar] [CrossRef]
- Handke, M.; Jastrzȩbski, W. Vibrational spectroscopy of the ring structures in silicates and siloxanes. J. Mol. Struct. 2004, 704, 63–69. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Shimojima, A.; Kuroda, K. Alkoxysilylated-Derivatives of Double-Four-Ring Silicate as Novel Building Blocks of Silica-Based Materials. Chem. Mater. 2008, 20, 1147–1153. [Google Scholar] [CrossRef]
- Nowacka, M.; Kowalewska, A.; Makowski, T. Structural studies on ladder phenylsilsesquioxane oligomers formed by polycondensation of cyclotetrasiloxanetetraols. Polymer 2016, 87, 81–89. [Google Scholar] [CrossRef]
- Blanco, I.; Bottino, F.A.; Abate, L. Influence of n-alkyl substituents on the thermal behaviour of Polyhedral Oligomeric Silsesquioxanes (POSSs) with different cage’s periphery. Thermochim. Acta 2016, 623, 50–57. [Google Scholar] [CrossRef]
- Vasil’ev, S.G.; Volkov, V.I.; Tatarinova, E.A.; Muzafarov, A.M. A Solid-State NMR Investigation of MQ Silicone Copolymers. Appl. Magn. Reson. 2013, 44, 1015–1025. [Google Scholar] [CrossRef]
- Uhlig, F.; Marsman, H.C. 29Si NMR—Some Practical Aspects. Silicon Compd. Silanes Silicones A Surv. Prop. Chem. 2008, 2, 208–222. [Google Scholar]
- Cheng, X.; Chen, D.; Liu, Y. Mechanisms of silicon alkoxide hydrolysis-oligomerization reactions: A DFT investigation. ChemPhysChem 2012, 13, 2392–2404. [Google Scholar] [CrossRef]
- Pierre, A.C. Introduction to Sol-Gel Processing; Springer Nature: Berlin, Germany, 2020; ISBN 9780792381211. [Google Scholar]
- Laird, M.; Yokoyama, J.; Carcel, C.; Unno, M.; Bartlett, J.R.; Wong Chi Man, M. Sol–gel processing of polyhedral oligomeric silsesquioxanes: Nanohybrid materials incorporating T8 and T10 cages. J. Sol-Gel Sci. Technol. 2020, 95, 760–770. [Google Scholar] [CrossRef]
- Wu, X.; Qin, Z.; Zhang, W.; Yang, R. KCl nanoparticles-loaded octaphenylsilsesquioxane as an efficient flame retardant for polycarbonate. React. Funct. Polym. 2022, 177, 105284. [Google Scholar] [CrossRef]
- Kamiya, K.; Dohkai, T.; Wada, M.; Hashimoto, T.; Matsuoka, J.; Nasu, H. X-ray diffraction of silica gels made by sol-gel method under different conditions. J. Non-Cryst. Solids 1998, 240, 202–211. [Google Scholar] [CrossRef]
- Jo, Y.Y.; Lee, A.S.; Baek, K.Y.; Lee, H.; Hwang, S.S. Multi-crosslinkable self-healing polysilsesquioxanes for the smart recovery of anti-scratch properties. Polymer 2017, 124, 78–87. [Google Scholar] [CrossRef]
- Garrido, J.; Linares-solano, A.; Martìn-Martìnez, J.M.; Molina-Sabio, M.; Rodrìguez-Reinoso, F.; Torregrosa, R. Use of N2 vs: CO2 in the Characterization of Activated Carbons. Langmuir 1987, 3, 76–81. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Kowalewska, A.; Fortuniak, W.; Handke, B. New hybrid silsesquioxane materials with sterically hindered carbosilane side groups. J. Organomet. Chem. 2009, 694, 1345–1353. [Google Scholar] [CrossRef]
- Moriones, P. Síntesis y Caracterización de Xerogeles Silíceos Híbridos (RTEOS/TEOS; R = P, Ph); Universidad Pública de Navarra: Pamplona, Spain, 2015; Available online: https://academica-e.unavarra.es/handle/2454/20351 (accessed on 13 October 2022).
- Kickelbick, G. Hybrid Materials Synthesis, Characterization, and Applications; John Wiley & Sons: New York, NY, USA, 2007; ISBN 9783527312993. [Google Scholar]
- Torres-Luna, J.A.; Carriazo, J.G. Porous aluminosilicic solids obtained by thermal-acid modification of a commercial kaolinite-type natural clay. Solid State Sci. 2019, 88, 29–35. [Google Scholar] [CrossRef]
- Sakka, S. Handbook of Sol-Gel Science and Technology: Processing, Characterization and Applications. Volume II: Characterization of Sol-Gel Materials and Products, 1st ed.; Sakka, S., Almeida, R.M., Eds.; Kluwer Academic Publishers: Osaka, Japan, 2005; ISBN 1-4020-7967-2. [Google Scholar]
- Rouquerol, J.; LLewelyn, P.; Rouquerol, F. Is the BET equation applicable to microporous adsorbents? Stud. Surf. Sci. Catal. 2007, 160, 49–56. [Google Scholar] [CrossRef]
- Rouquerol, F.; Rouquerol, J.; Sing, K.S.W.; Llewellyn, P.; Maurin, G. Adsorption by Powders and Porous Solids Principles, Methodology and Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 1999; ISBN 978-0-12-598920-6. [Google Scholar]
- García-Martínez, J.; Cazorla-Amorós, D.; Linares-Solano, A. Further evidences of the usefulness of CO2 adsorption to characterise microporous solids. Stud. Surf. Sci. Catal. 2000, 128, 485–494. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | Wavelength | Vibration | Structural | Reference |
|---|---|---|---|---|
| (cm−1) | Assignation | Unit | ||
| 1 | 1380 | ν (C=C) | Si–Ph–Cl | [36,48] |
| 2 | 1190 | νas (Si–O–Si), LO mode | (SiO)6 | [40,44] |
| 3 | 1160 | νring-as (Si–O–Si), LO mode | (SiO)4; T7 | [49] |
| 4 | 1135 | νring-as (Si–O–Si), LO mode | (SiO)4; T8 | [40,44,49,50,51] |
| 5 | 1120 | νring-s (Si–O–Si), LO mode | (SiO)4; T7, Sld | [49] |
| 6 | 1085 | νas (C=C) | Si–Ph–Cl | [48] |
| 7 | 1050 | νas (Si–O–Si), TO mode | (SiO)4, Sld | [40,44,49,51] |
| 8 | 1030 | νas (Si–O–Si), TO mode | (SiO)6 | [40,44] |
| 9 | 1015 | νs (C=C) | Si–Ph–Cl | [48] |
| 10 | 1005 | νas (Si–O–Si) | Lineal siloxane | [50] |
| 11 | 950 | ν (Si-O−/Si-OH) | Si–OH | [40,44,46] |
| 12 | 815 | Τδ,y C–H | Si–Ph–Cl | [36,48] |
| 13 | 760 | Φ C–H | Si–Ph–Cl | [36] |
| 14 | 710 | C–Cl | Si–Ph–Cl | [36,47] |
| Hybrid Material | LO6 | LO4 | TO4 | TO6 | (SiO)4 | (SiO)6 |
|---|---|---|---|---|---|---|
| (%) | (%) | |||||
| 0ClPh | 7.0 | 28.5 | 18.1 | 46.5 | 46.5 | 53.5 |
| 5ClPh | 26.7 | 31.8 | 38.6 | 3.0 | 70.4 | 29.6 |
| 10ClPh | 12.8 | 43.9 | 29.8 | 13.6 | 73.7 | 26.3 |
| 15ClPh | 12.3 | 43.1 | 33.9 | 10.7 | 77.0 | 23.0 |
| 20ClPh (Monolith) | 3.3 | 13.4 | 30.9 | 52.4 | 44.3 | 55.7 |
| 20ClPh (Precipitate) | 2.6 | 21.0 | 68.3 | 8.1 | 89.6 | 10.8 |
| 100ClPh | 0.4 | 77.2 | 20.2 | 2.3 | 97.4 | 2.7 |
| Xerogel | 29Si NMR (ppm) | Band Areas | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T2 | T3 | Q2 | Q3 | Q4 | T | T2 | T3 | T3/T2 | |
| 0ClPh | a | a | −92.1 | −100.9 | −109.0 | a | a | a | - |
| 1ClPh | a | a | −92.3 | −101.0 | −109.3 | a | a | a | - |
| 5ClPh | −69.8 | −78.7 | −92.3 | −101.1 | −109.4 | 5.5 | 4.7 | 0.8 | 0.2 |
| 10ClPh | −69.8 | −79.1 | −92.2 | −101.0 | −108.5 | 12.8 | 8.4 | 4.4 | 0.5 |
| 15ClPh | −69.8 | −78.2 | −92.1 | −100.9 | −108.6 | 17.6 | 11.1 | 6.5 | 0.6 |
| Xerogel | Peak 2θ < 10° | Peak 10° > 2θ < 30° | ||||
|---|---|---|---|---|---|---|
| 2θ1 (°) | A1 | d1 (nm) | 2θ2 (°) | A2 | d2 (nm) | |
| 0ClPh | a | a | a | 24.11 | 6481 | 0.369 |
| 1ClPh | a | a | a | 23.93 | 6845 | 0.372 |
| 5ClPh | a | a | a | 23.58 | 5921 | 0.377 |
| 10ClPh | 3.6 | 2112 | 2.43 | 24.14 | 5778 | 0.369 |
| 15ClPh | 4.1 | 1985 | 2.17 | 24.53 | 5534 | 0.363 |
| Xerogel | aBET | aDR | Vmicro | Vmicro | Vmeso | Vtotal | BJH APS a | Ec b | Ec b |
|---|---|---|---|---|---|---|---|---|---|
| (N2) | (CO2) | (N2) | (CO2) | (N2) | (N2) | (N2) | (CO2) | ||
| (m2 g−1) | (cm3 g−1) | (nm) | (KJ mol−1) | ||||||
| 0ClPh | 697 | 510 | 0.283 | 0.195 | 0.074 | 0.407 | 3.61 | 15.27 | 19.71 |
| 1ClPh | 656 | 426 | 0.253 | 0.163 | 0.222 | 0.557 | 4.38 | 15.57 | 19.97 |
| 3.5ClPh | 504 | 429 | 0.205 | 0.164 | 0.007 | 0.223 | 3.33 | 18.73 | 19.28 |
| 5ClPh | 493 | 400 | 0.205 | 0.153 | 0.006 | 0.209 | 3.40 | 18.11 | 19.77 |
| 7.5Clph | 431 | 388 | 0.176 | 0.148 | 0.004 | 0.177 | 3.29 | 19.49 | 18.92 |
| 10ClPh | 367 | 363 | 0.151 | 0.139 | 0.003 | 0.147 | 2.05 | 16.24 | 19.29 |
| 15ClPh | 497 | 358 | 0.208 | 0.137 | 0.007 | 0.212 | 3.11 | 15.61 | 19.55 |
| Xerogels | Weight Percentage of Chlorine | E/T | |
|---|---|---|---|
| (wt%) | |||
| Theoretical (T) | Experimental (E) | ||
| 1ClPh | 0.37 | 0.29 | 0.77 |
| 5ClPh | 1.68 | 1.20 | 0.72 |
| 10ClPh | 2.97 | 2.80 | 0.94 |
| 15ClPh | 3.99 | 2.28 | 0.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Quesada, G.; Espinal-Viguri, M.; López-Ramón, M.V.; Garrido, J.J. Novel Silica Hybrid Xerogels Prepared by Co-Condensation of TEOS and ClPhTEOS: A Chemical and Morphological Study. Gels 2022, 8, 677. https://doi.org/10.3390/gels8100677
Cruz-Quesada G, Espinal-Viguri M, López-Ramón MV, Garrido JJ. Novel Silica Hybrid Xerogels Prepared by Co-Condensation of TEOS and ClPhTEOS: A Chemical and Morphological Study. Gels. 2022; 8(10):677. https://doi.org/10.3390/gels8100677
Chicago/Turabian StyleCruz-Quesada, Guillermo, Maialen Espinal-Viguri, María Victoria López-Ramón, and Julián J. Garrido. 2022. "Novel Silica Hybrid Xerogels Prepared by Co-Condensation of TEOS and ClPhTEOS: A Chemical and Morphological Study" Gels 8, no. 10: 677. https://doi.org/10.3390/gels8100677
APA StyleCruz-Quesada, G., Espinal-Viguri, M., López-Ramón, M. V., & Garrido, J. J. (2022). Novel Silica Hybrid Xerogels Prepared by Co-Condensation of TEOS and ClPhTEOS: A Chemical and Morphological Study. Gels, 8(10), 677. https://doi.org/10.3390/gels8100677