Abstract
We present results of MD and MC simulations of the equilibrium properties of swelling gels with comb-like or bottlebrush subchains and compare them to scaling-theory predictions. In accordance with theory, the simulation results demonstrate that swelling coefficient of the gel increases as a function of the polymerization degree of the main chains and exhibits a very weak maximum (or is virtually constant) as a function of the polymerization degree and grafting density of side chains. The bulk osmotic modulus passes through a shallow minimum as the polymerization degree of the side chains increases. This minimum is attributed to the onset of overlap of side chains belonging to different bottlebrush strands in the swollen gel.
1. Introduction
Brush-like macromolecules (molecular brushes) have been extensively studied theoretically and experimentally for a number of decades [1,2,3,4,5,6,7,8,9,10,11]. Both intra- and intermolecular interactions between side chains densely attached to the molecular backbone determine specific conformational and dynamic properties of molecular brushes, as compared to linear analogues. Chemical (covalent) cross-linking of brush-like polymers with subsequent swelling in a good solvent gives rise to the so called “hairy” gels with strands constituted by molecular brushes. The swelling ratios and osmotic moduli of such gels depend in a complex way on the grafting density and polymerization degree of the side chains decorating the network strands [12,13,14,15,16]. Similar structures (“hairy mesogels”) arise upon self-assembly of triblock copolymers with a comb-like or bottlebrush central block and associated terminal blocks [12,17]. In these, physically cross-linked networks the strands are formed by central blocks of copolymer (molecular brushes) that connect neighboring domains of associated blocks (network cross-links).
A molecular brush consists of a linear chain backbone with multiple side chains tethered to it. Three major synthetic approaches: (i) “grafting to” (pre-synthesized side chains are covalently attached to the backbone); (ii) “grafting through” (polymerization of the so-called macromonomers), and (iii) “grafting from” (side chains are polymerized from the backbone as macroinitiator) produced a wide variety of molecular brushes with linear grafts. An additional structural complexity can be introduced through selective gradients in grafting density or block copolymers as side chains [2,18,19].
The possibility to vary architectural parameters of molecular brushes (such as side chain length and grafting density) allows for control and adjustment of static and dynamic properties of both bulk materials and gels thereof [2,6]. Furthermore, branched architectures with bottlebrush motifs, such as barbwire [20,21,22] or dendronized polymers [23,24], have been synthesized and explored theoretically and experimentally [25,26,27]. Combining branched architecture with temperature-, pH-, and light-responsive functions in the main and side chains of molecular brushes opens new opportunities for smart material design [17,28,29,30,31,32,33,34,35]. Since properties of the constituent macromolecules play a governing role in functions of the materials, analytical theories [36,37,38,39,40,41,42] and self-consistent field numerical [43] approaches; and coarse-grained computer simulations [44,45,46,47,48,49,50,51,52,53] of branched polymers have been used to corroborate the relationships between macromolecular architecture and experimentally accessible properties of melts, solutions, and thin films comprising molecular brushes.
It was demonstrated [31] that thermo-responsive triblock copolymers with linear (L) terminal and bottlebrush (B) central blocks can produce hydrogels upon association of L-blocks in spherical domains physically cross-linking B-strands. For example, PNIPAM-bbPEG-PNIPAM triblocks self-assembled upon reaching their lower critical solution temperature (LCST) to produce two types of polymer networks: injectable hydrogels at body temperature and elastomers after water evaporation [31]. The gelation process was attributed to LCST-triggered microphase separation of the PNIPAM L-blocks, and the forming network in both hydrogels and elastomers was homogeneous, in contrast to microphase-separated linear counterparts.
Self-assembly of bottlebrush block copolymers in solutions remains a topic of intensive experimental [54,55,56,57,58,59,60,61,62,63,64] and theoretical [42,65,66] research. It was demonstrated that branched architecture of soluble blocks leads to a variety of self-assembling aggregates that could potentially serve as precursors of gels with bottlebrush strands.
The goal of this paper is to compare the recent theoretical predictions on “hairy” gels with the results of MD and MC computer simulations. In Section 2, we briefly review the scaling model of hairy gels, and summarize the theoretical predictions on its swelling behavior and mechanical properties. We then compare the theoretical predictions with the data from Monte Carlo (MC) and molecular dynamic (MD) simulations. In Section 4, we present the details of implemented MC and MD methods. In Section 3, we formulate conclusions and outline perspectives for further development in theory and modeling of bottlebrush architectures.
2. Results and Discussion
2.1. “Hairy” Polymer Gel: Scaling Model
Swelling of chemically and/or physically cross-linked networks with brush-like strands in a good solvent yields “hairy” gels. Each strand in a hairy gel constitutes a molecular brush with degree of polymerization (DP) M in its flexible backbone (main chain), and equally flexible spacers and side chains with DPs m and n, respectively (see Figure 1a). In contrast to networks with linear strands, the swelling and elastic properties of hairy gels depend not only on the cross-linking density defined by the strand DP , but also on n and grafting density () of side chains at a given M. If , side chains overlap and stretch normally to the backbone due to monomer–monomer interactions, giving rise to a bottlebrush (molecular brush). If , the side chains exhibit coil conformations, making the polymer comb-like.
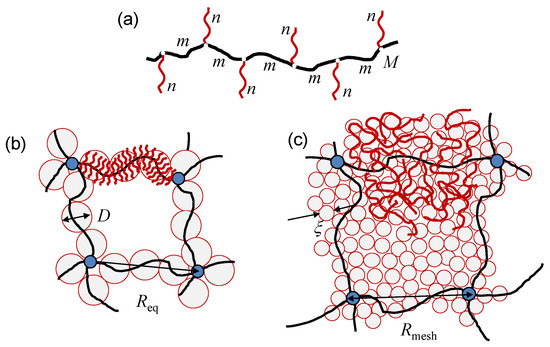
Figure 1.
Schematics of graft-polymer, with being the DPs of the main and side chains and of the spacers, respectively. (a) Hairy gel with bottlebrush strands in hollow mesh (b) and filled mesh (c) regimes. Backbones of strands are colored in black, side chains are in red, and cross-links are marked as blue circles. is the equilibrium end-to-end distance of the main chains of the gel strands (the mesh size). Superblobs with size D (hollow-mesh regime) and concentration blobs with size in semi-dilute solution of side chains (filled-mesh regime) are shaded light gray.
A molecular brush is envisioned as a wormlike chain with thickness D (which is end-to-end distance of the side chains normally to the backbone), spacer end-to-end distance h, effective contour length , and persistence length . In the framework of scaling model [36,42], the strand thickness D and spacer end-to-end distance h are specified by the balance between the elastic stretching of side chains and spacers, and repulsive monomer-monomer interactions in the cylindrical layer around the main chain, to give
The first and the second lines in Equations (1) and (2) correspond to partial and close to maximal stretching of spacers in the main chain, respectively (with separating two elasticity regimes for the backbone); a is the length of monomer unit in the backbone and side chain; and is Flory exponent ( in good solvent, and in theta-solvent). Notably, the scaling relations in Equations (1) and (2) have asymptotic character—that is, they apply only for , , and with
In scaling terms, (or, equivalently ) separates bottlebrushes with from starlike polymers with . In realistic experimental and simulation systems, and . Due to the local cylindrical symmetry, a strong overlap of moderately long side chains occurs only close to the backbone, and therefore the scaling dependences predicted by Equations (1) and (2) are only approached in the currently attainable range of n, and M. However, due to still noticeable stretching of the side chains normally to the backbone (), a bottlebrush molecule behaves as a self-avoiding chain composed of impermeable subunits (“superblobs” with size D each), and its overall size in dilute solution scales as
Distribution of polymer’s density within a swollen hairy gel could be inhomogenuous. Two structural regimes, hollow mesh and filled mesh gel, are distinguished depending on the ratio between the mesh size, , and the superblob size, D [15,16]. In the hollow mesh regime (Figure 1b), each strand constitutes a molecular brush with thickness , and weak overlap of neighboring strands ensures hollow space (mesh) between cross-links. In this case, the mesh size can be evaluated using the -theorem of de Gennes, [67] to give . In the filled-mesh regime (Figure 1c), the neighboring strands strongly overlap, giving rise to a semi-dilute solution of side chains with an almost uniform concentration . The interior of the gel is envisioned as a closely-packed array of the concentration blobs with size . In this case, the equilibrium mesh size results from the balance between gel osmotic pressure, , and the conformational elasticity of the backbones (renormalized according to the polymer concentration c inside the gel).
To facilitate comparison between the theoretical predictions and the simulation data, we present in Figure 2 the scaling-type diagram of states for hairy gels [16] in log–log coordinates (with ). In the hollow-mesh regimes and , each spacer in the main chain is, respectively, partially or almost fully stretched. The boundary corresponds thereby to , indicating the onset of spacer strong stretching in individual strands at given Similar situation occurs in the filled mesh regimes and . That is, the boundary corresponds to the onset of spacer strong stretching in the filled-mesh regimes (semi-dilute solutions of the side chains). At the boundaries and (red lines in Figure 2), DP M of the backbone becomes equal (in scaling terms) to , separating bottlebrush and starlike conformations of strands. At these boundaries (indicated by ), each strand comprises on the order of one superblob with size D (), as schematically shown in Figure 2.
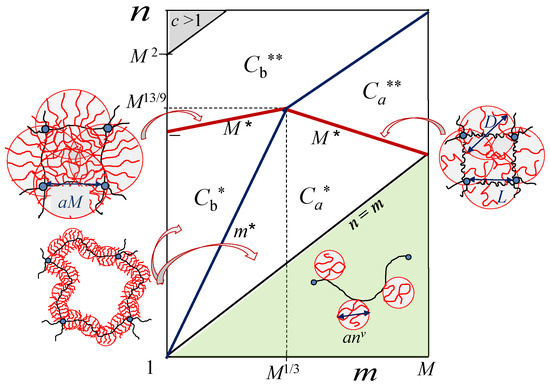
Figure 2.
Scaling-type diagram of states for hairy gel in coordinates in good solvent (). In the hollow-mesh regimes and , bottlebrush strands have either partially or almost fully stretched spacers, respectively. In filled-mesh regimes and , the interior of the gel constitutes a semi-dilute solution of side chains. Schematics demonstrate strand conformations in regimes and , and at boundaries and (that correspond to backbone length at which bottlebrush transforms in starlike polymer with ). Boundary (marked ) separates bottlebrush strands with fully () and partically () stretched spacers. In the shaded green area, strands are comb-like (side chains are unstretched coils). Strands in regimes of semi-dilute solutions and are shown in Figure 1.
The scaling expressions for the equilibrium mesh size , gel swelling coefficient (that is, ratio of volumes V in the swollen and dry states)
with and , and osmotic bulk modulus
are collected in Table 1 with corresponding exponents specified for .

Table 1.
Asymptotic power-law dependencies for normalized mesh size , swelling ratio Q, and bulk osmotic modulus G in a free-swelling hairy gel with DP M of strands in good solvent ().
As follows from Table 1, an increase in DP n of the side chains at fixed DP M of the backbone leads to the monotonous increase in mesh size, , in regimes and (exponent , (exponent ), approaching full extension, , in regime (exponent ). At the same time, both swelling ratio Q and osmotic bulk modulus G exhibit non-monotonic dependences on n. Swelling coefficient passes through a maximum at the boundaries and , and osmotic modulus passes through a minimum upon crossing the boundaries of regime . The maximum in dependence is weak due to a small value of the exponent which changes from to at the boundary, and this maximum could even disappear if the apparent exponent . Recall that the values of exponents in Table 1 were calculated in the limit of . The predicted sharp (jumpwise) minimum in at the boundaries of regime could be smoothed in computer simulations, leading to shift in the minimum location far left (to smaller values of n).
2.2. Computer Simulations of Hairy Gel
We used both MC and MD simulations to explore the equilibrium-swelling behavior of hairy gels in a good solvent (). In Figure 3, we present the simulation box which has the same geometry in MC and MD simulations. That is, 16 polymer chains were connected to a diamond-like network by eight tetrafunctional cross-linking units, and the network element was put into cubic simulation box of the volume V with periodic boundary conditions to emulate an infinite polymer network. It is worth mentioning that MD and MC simulations were performed in and (with pressure and V fluctuating) ensembles, respectively.
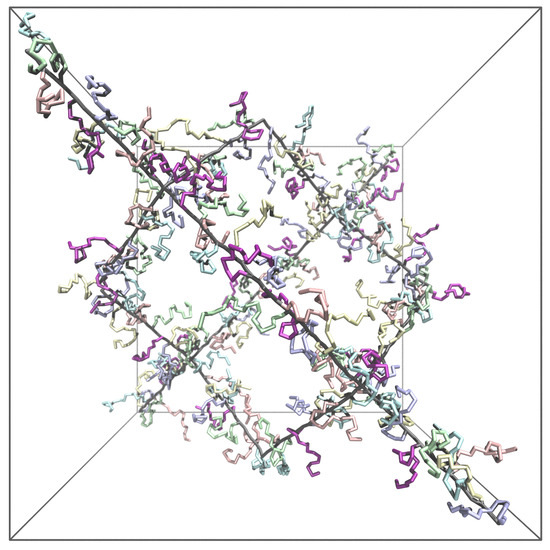
Figure 3.
A view of a simulation unit cell with gel backbone (in black color, length ) and side chains (in various light colors for clarity, length ) grafted every second monomer unit () intentionally in a state with pressure P < 0 (volume V greater than in free swelling equilibrium with pressure ) to avoid excessive overlapping of side-chains and clearly showing the gel branching. Backbones are indicated in black with side chains in various colors (to avoid crowding).
In Figure 4a,b, we present the scaling-type diagrams with positions of the boundaries calculated specifically for DP and DP of the backbone, respectively, together with set of symbols marking the architectural parameters of bottlebrush strands modeled in MC simulations. In Figure 4c, a similar diagram is presented for with set of symbols marking the parameters of strands modeled in MD simulations.
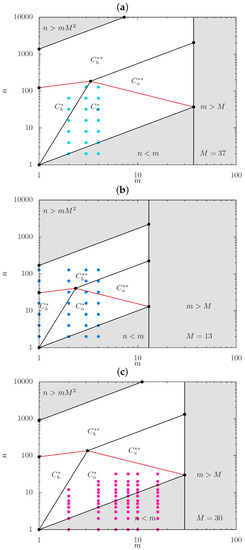
Figure 4.
Scaling-type diagrams with positions of boundaries calculated specifically for DP (a) and DP (b) of the backbone, respectively, together with ordered set of symbols marking the architectural parameters of bottlebrush strands modeled in MC simulations and (c) for in MD simulations.Shaded area with corresponds to unphysical values of .
While positions of the boundaries in Figure 4 are specified with accuracy of the numerical prefactors on the order of unity, it is still expected that MC simulations for (Figure 4a) with small values of and increasing n (up to ) cover regimes and . In contrast, Figure 4b indicates that for , MC data pass through regimes , and even enters regime . According to Figure 4c, MD simulations cover a considerably larger interval of m-values (up to ). However, many of the symbols correspond to comb-like strands (area with shaded gray), and the rest of the data probes only regime .
2.2.1. Average End-to-End Distances of Strands and Side Chains
In Figure 5, we plot the average mesh size of a free-swelling gel, and the average end-to-end distance of the side chains obtained in MC simulations as a function of n for and 4 in log–log coordinates.
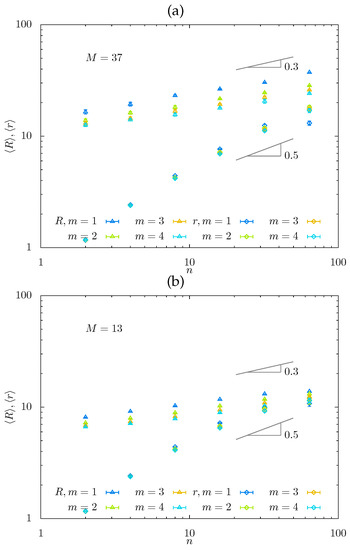
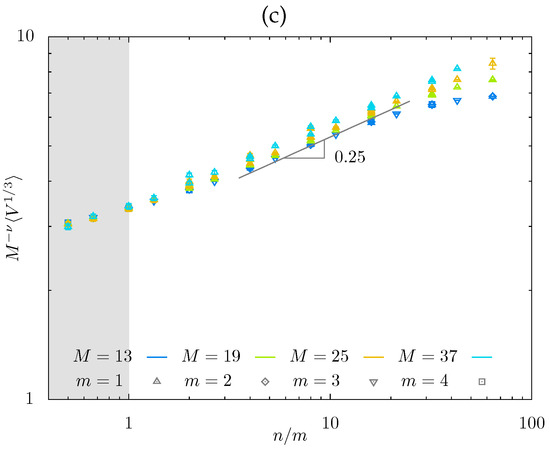
Figure 5.
Average mesh size and end-to-end distances of the side chains as a function of n for for (a) and (b) obtained from MC simulations. In panel (c), the mesh size is normalized by . Here and below, the gray area corresponds to comb-like strands with .
At (when the side chains stretch normally to the backbone), different m-values produce almost parallel lines in dependences, with apparent slopes , i.e., smaller than the theoretically predicted (i.e., in regime and in regime ). For , the correspondence between the theoretical exponents (i.e., in regimes and in regime ) and apparent slopes is expectably [50] worse, consistent with smaller apparent values of . Notably, at , , and do not intersect at fixed m at any considered n (see Figure 5a), indicating that the hairy gel remains in the hollow mesh state. In contrast, at , and become close at the largest values of n (see Figure 5b), indicating that the hairy gel approaches the filled-mesh state. In Figure 5c, we plot normalized mesh size as a function of ( for , and 37 to demonstrate how MC data collapse on mastercurve with slope , with maximal deviations for the smallest value of (presumably approaching regime with ).
In Figure 6, the equilibrium strand end-to-end distance is presented for a series of m-values and fixed as obtained from MD simulations. While values of correspond to comb-like strands, the data for relatively small m and are used in Figure 6 to evaluate apparent exponent in the dependence to give , in accordance with the results of MC simulations.
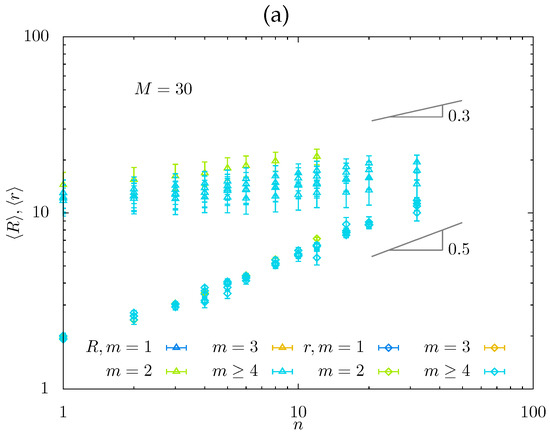
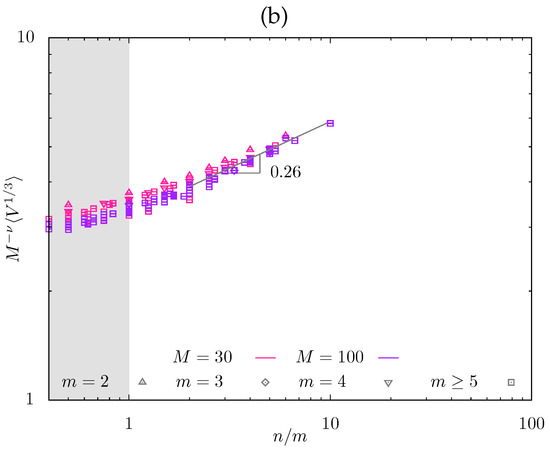
Figure 6.
Equilibrium mesh size from MD simulations, , as a function of n for a series of m-values and fixed (a), and normalized mesh size versus with slope (b).
2.2.2. Gel Swelling Coefficient
In Figure 7, we present the normalized equilibrium swelling coefficient as a function of predicted by the scaling model (Figure 7a) and obtained from MC simulations (Figure 7b) in log–log coordinates. The power-law dependences in Figure 7a were calculated for with slopes indicated in Table 1 (that is, and in regimes , respectively) and , with all numerical prefactors assigned unity. For , Q is shown by red lines; for only regimes are feasible. The maximum predicted for corresponds to . A decrease in M shifts location of the maximum to the left and makes it less pronounced.
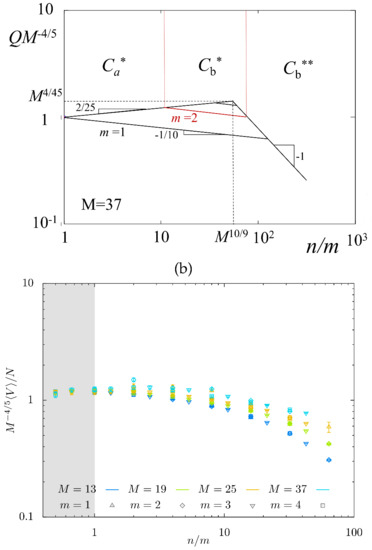
Figure 7.
Theoretical (a) and MC-simulated (b) swelling coefficient Q as a function of . Scaling dependences for theoretical Q (with all numerical coefficients equal to unity) were calculated for . and boundaries are indicated for .
MC simulations data in Figure 7b show the dependence of on for series of M-values. The predicted weak maximum is not well pronounced; mostly, its decreasing (right) branch is seen in MC simulations. As strands with are not expected to enter regime (see Figure 4a), the slope is far from the predicted exponent . However, for smaller for which regime is feasible (see Figure 4b), the slope of MC data approaches , as predicted.
The data from MD simulations in Figure 8 also indicate weak dependence of swelling coefficient Q on . Here, the normalized swelling coefficient is presented as a function of for a wider variety of m-values and two values of (red symbols) and (violet symbols), and probing regime in which the predicted slope is For , the data in Figure 8 with different m-values remain rather scattered; the increase in backbone DP M up to decreases the scattering of the data that collapse on mastercurve with close to zero slope. Notably, the numerical prefactor in versus dependences is close to unity in both MC and MD simulations, consistent with the scaling model.
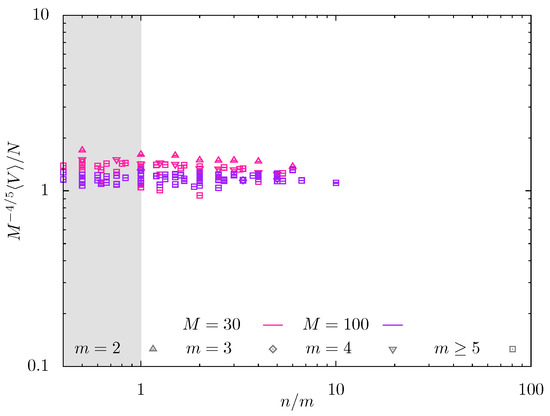
Figure 8.
Normalized swelling coefficient from MD simulations for (red symbols) and 100 (violet symbols) as a function of .
2.2.3. Osmotic Bulk Modulus
In Figure 9a we present the MC data for the gel osmotic bulk modulus, . Here, is osmotic pressure, and is the equilibrium concentration of monomer units in the hairy gel. As is seen in Figure 9a, the osmotic modulus G decreases with increasing DP M of the strand backbone, in qualitative agreement with the theoretical predictions in Table 1. However, the predicted minimum in dependence is not detected in MC simulations. An increase in M flattens dependence, only pointing at the possibility of minimum formation.
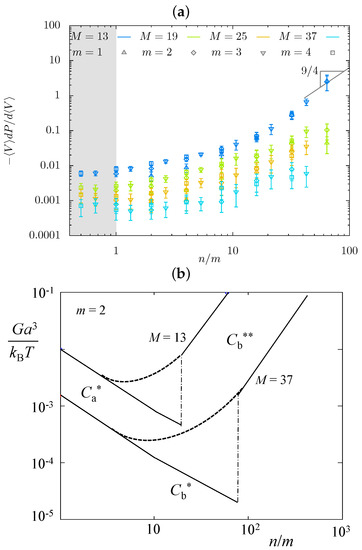
Figure 9.
Osmotic modulus G as a function of in MC simulations (a) and scaling model (b). A conjecture that sharp minimum could be smoothed in simulations is illustrated by the dashed lines (b).
A conjecture is that the sharp minimum predicted by the scaling model could be smoothed in MC simulations, as schematically illustrated by the dashed lines in Figure 9b. Another factor is the effect of perturbed dense cylindrical layers circumventing the backbones. Although Figure 9b is merely schematic and does not specify the shape or position of the dotted lines, it is clear that the theoretically predicted exponents for would not work in the smoothed region, and therefore, collapse of G-data on the mastercurve is not expected. However, this schematic indicates that minimum in dependence can move left and fall out of the considered ranges of the strand architectural parameters.
3. Conclusions
Based on the scaling description of molecular brushes, the theory specifies power-law dependences for the equilibrium properties of “hairy” gels with bottlebrush strands. Four structural regimes of hairy gels were distinguished: two hollow-mesh regimes, and , with partially and almost fully stretched spacers; and two filled mesh regimes, and , with partially or almost fully stretched backbones embedded in a semi-dilute solution of side chains. Two supplementary computer simulation techniques, MC and MD, were used to probe the structure of free-swelling in good solvent gel with varied strand architectural parameters to corroborate the theoretical model. It was demonstrated that MC simulations of hairy gels with short spacers () could cover regimes and , and approach regime . MD simulations of gels with a wider variety of m-values could probe regime . In all cases, limited extension of the gel regimes (Figure 4) and restriction make it challenging to compare the predicted asymptotic values of exponents (Table 1) with apparent exponents from MC and MD simulations. In addition, it is not surprising that apparent exponents deviate from the theoretical ones for the considered set of strand parameters .
According to the scaling model, the gel mesh size increases monotonously with increasing of both the DP n of the side chain and the DP M of the strand backbone. The apparent exponent estimated from MC and MD simulations was smaller but reasonably close to the theoretical values, . Exponent was close to as expected for good solvent conditions. The thickness D of the bottlebrush strand increased with n. However, an approach to the theoretical exponent requires (i.e., much larger n-values than have been implemented in MC simulations), and the correspondence between apparent and scaling exponents in dependence was poor.
The theory predicts that the swelling ratio Q of the hairy gels is controlled primarily by the DP M of the strand backbone, whereas the dependence on the DP n and grafting density of the side chains is weak. This prediction is in agreement with the MC and MD simulation data. The theory also predicts non-monotonic dependences of osmotic modulus G and swelling ratio Q of hairy gels on DP n of the side chains. The osmotic modulus G passes through a minimum corresponding to the overlap threshold of the side chains emanating from different strands. In contrast, swelling ratio Q passes through a maximum upon onset of strong stretching of spacers in nonoverlaping strands. The MC computer simulations did not find pronounced extrema in and dependences and demonstrated only the decreasing branch in Q-dependence (with slope approaching , predicted in regime ) and the increasing branch in G-dependence (with slope approaching , predicted in regime ). Lack of maximum in dependence can be explained by the values of apparent exponent in dependence that automatically eliminates the increasing branch in . The relatively small values of could follow from the limited extension of the scaling regimes for currently considered set of parameters. A wider set of architectural parameters is desirable to confront the scaling model of hairy gel with better accuracy.
4. Materials and Methods
The model of the gel as a network of 16 linear polymer chains, each consisting of M monomer units. These polymer chains are connected to a diamond-like network by eight cross-linking units. The side chains of the lenght n are grafted on each m-th monomer of the main chain (see Figure 3). The network was put into cubic simulation box of the volume with periodic boundary conditions, which virtually emulates an infinite polymer network.
4.1. Monte Carlo Simulations
In Monte Carlo simulations, we used a variant of the Hamiltonian (originally called hybrid [68]) Monte Carlo (HMC) method and coarse-grained (CG) models. The HMC uses Hamiltonian dynamics to sample probability distribution , where is the Hamiltonian, q are generalized coordinates, and p are generalized momenta. In the simplest case, a separable Hamiltonian is used, where K is kinetic energy and V is potential energy for sampling Boltzmann distribution . The momenta p can be sampled directly, and Hamiltonian dynamics , are followed for some time to prepare a new proposal for a Metropolis step [69] with acceptance probability . For the exact dynamics , it would be zero (time independent Hamiltonian is conserved), and all proposals would be accepted. In practice, instead of exact dynamics, we must use an approximate numerical evolution with numerical integrators. Fortunately, time-reversible, phase-space volume-preserving integrators are available, and their procedure is exact, and any bias due to approximate dynamics is then removed by Metropolis rejections. Moreover, the Hamiltonian for dynamic evolution can be different from the targeted one. This allows for great variability of the method. In the case of polymer simulations, a big improvement can be achieved by a suitable transformation of variables. The potential energy for the standard Rouse (harmonic) model is diagonal in bond vector coordinates . With the freedom of choosing the evolution Hamiltonian, we use new canonically conjugated momenta with bond vector coordinates, and it allows for sampling all normal modes at the same rate [70]. For other potentials and approximate integrators, the method somewhat deteriorates, but it is still much better than simple molecular dynamics and leads to a smaller scaling exponent of the integrated autocorrelation time of the end-to-end distance with chain length, and thus is arbitrarily faster.
In our CG model we use two types of interactions, bonding and non-bonding. For the bonding one, we use a variant of finitely extensible nonlinear elastic (FENE) potential [71] in a form for and infinite elsewhere, with mild singularities at . It is a relatively small complication for simple molecular dynamics where small time-steps are used that potential does not exist outside . In HMC, we use the advantage of much longer time-steps that could lead to stepping out of the definition interval. We resolve this in the spirit of HMC and prepare a new potential finite, everywhere being on and , where elsewhere that is used for dynamic evolution in HMC and the original is used for the Metropolis step. By this combination, we achieve the exact sampling with while avoiding numerical problems. For non-bonding potential, we use soft repulsive potential for and 0 elsewhere, where is an energy parameter and c is a cutoff. The potential is smooth at cutoff. Frequently, the standard Lennard–Jones potential [72], originally suggested for simulations of neon, is used for this purpose. Although the part can be justified for two atoms (with at least one in an S-state) at very long distances, the part lacks such a simple justification, the less for the potential that should represent CG macromolecular system in a solution. By taking a systematically coarse-grained model based on all-atom empirical force-field and fitting the corresponding part for small distances, we find a much smaller exponent [73] of about 2, as for our .
The simulations were performed in ensemble by adding volume-changing moves [74]. The simulations started from extended conformations of gel backbone and coiled side-chains as corresponding to a good solvent. A thorough equlibration was performed for box sizes, energies, and end-to-end distances. The free (tuning) parameters of our HMC method were roughly set up according to optimal acceptance probability and the smallest integrated autocorrelation times. After equilibration (as illustrated by Figure 10), samples were accumulated, and standard deviations of their averages were estimated using the blocking method [75].
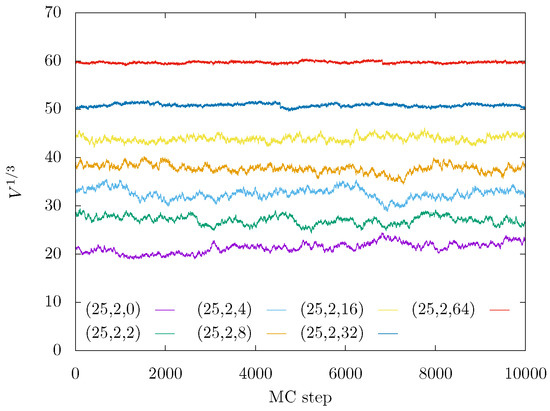
Figure 10.
Traces of instantaneous box sizes during NPT Monte Carlo sampling for and gel architectural parameters , as indicated in the legend.
4.2. Molecular Dynamics Simulations
Each pair of the particles interact via the truncated Lennard–Jones interaction potential, which imposes strong repulsion between all particles at short distances:
where r is the interparticle distance, nm is a chosen characteristic size of the particles, is the depth of the potential, and is the cut-off distance beyond which the potential is set zero.
The bonds connecting the gel to a network are modeled using finite extension nonlinear elastic potential (FENE):
where r is the distance between the bonded segments, K is the magnitude of their interaction, is the maximal stretching length of the bond, and is the equilibrium bond length. In our simulations we set , and [76].
The Langevin thermostat [71] was used to guarantee the constant temperature of the system, K. The two additional terms to force in equation of motion were added.
where the first term corresponds to constant friction, with being a friction coefficient, and the second one corresponds to random thermal force, with being a normally distributed random vector; —velocity of i-th particle, t—time.
In order to calculate the swelling ratio of hydrogel, we performed a series of simulations of the gel in a box of different volumes V. Each simulation results in a particular value of pressure P.
Our target observable is the free swelling equilibrium state, i.e., at the state where the applied to the gel pressure equals to zero. In order to localize this state, we first plotted dependence; then, using the least-squares method, we drew a line passing through the nearest to the V axis points. The place where this line crosses the V-axis defines the volume of free swelling equilibrium state.
Author Contributions
Scaling theory O.V.B. and E.B.Z.; MC simulations F.U.; MD simulations O.V.R.; writing all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Russian Science Foundation, grant 20-13-00270.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
There are no conflict of interest to declare.
References
- Sheiko, S.S.; Sumerlin, B.S.; Matyjaszewski, K. Cylindrical molecular brushes: Synthesis, characterization and properties. Prog. Polym. Sci. 2008, 33, 759–785. [Google Scholar] [CrossRef]
- Verduzco, R.; Li, X.; Pesek, S.L.; Stein, G.E. Structure, function, self-assembly of bottlebrush copolymers. Chem. Soc. Rev. 2015, 44, 2405–2420. [Google Scholar] [CrossRef] [PubMed]
- Müllner, M.; Müller, A.H.E. Cylindrical polymer brushes—Anisotropic building blocks, unimolecular templates and particulate nanocarriers. Polymer 2016, 98, 389–401. [Google Scholar] [CrossRef]
- Rzayev, J. Molecular Bottlebrushes: New Opportunities in Nanomaterials Fabrication. ACS Macro Lett. 2012, 1, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Prukop, S.L.; Biswal, S.L.; Verduzco, R. Surface Properties of Bottlebrush Polymer Thin Films. Macromolecules 2012, 45, 7118–7127. [Google Scholar] [CrossRef]
- Liang, H.; Sheiko, S.S.; Dobrynin, A.V. Supersoft Polymer Networks with Brushlike Strands. Macromolecules 2018, 51, 638–645. [Google Scholar] [CrossRef]
- Yuan, J.; Müller, A.H.E.; Matyjaszewski, K.; Sheiko, S. Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Clair, C.; Lallam, A.; Rosenthal, M.; Sztucki, M.; Vatankhah-Varnosfaderani, M.; Keith, A.N.; Cong, Y.; Liang, H.; Dobrynin, A.V.; Sheiko, S.S.; et al. Strained Bottlebrushes in Super-Soft Physical Networks. ACS Macro Lett. 2019, 8, 530–534. [Google Scholar] [CrossRef]
- Xie, G.; Martinez, M.R.; Olszewski, M.; Sheiko, S.S.; Matyjaszewski, K. Molecular Bottlebrushes as Novel Materials. Biomacromolecules 2019, 20, 27–54. [Google Scholar] [CrossRef]
- Rathgeber, S.; Pakula, T.; Wilk, A.; Matyjaszewski, K.; Beers, K.L. On the shape of bottle-brush macromolecules: Systematic variation of architectural parameters. J. Chem. Phys. 2005, 122, 124904. [Google Scholar] [CrossRef]
- Li, Z.; Tang, M.; Liang, S.; Zhang, M.; Biesold, G.M.; He, Y.; Hao, S.-M.; Choi, W.; Liu, Y.; Peng, J.; et al. Bottlebrush polymers: From controlled synthesis, self-assembly, properies to applications. Prog. Polym. Sci. 2021, 116, 101397. [Google Scholar] [CrossRef]
- Li, T.; Huang, F.; Diaz-Dussan, D.; Zhao, J.; Srinivas, S.; Narain, R.; Tian, W.; Hao, X. Preparation and Characterization of Thermoresponsive PEG-Based Injectable Hydrogels and Their Application for 3D Cell Culture. Biomacromolecules 2020, 21, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Sarapas, J.M.; Chan, E.P.; Rettner, E.M.; Beers, K.L. Compressing and Swelling To Study the Structure of Extremely Soft Bottlebrush Networks Prepared by ROMP. Macromolecules 2018, 51, 2359–2366. [Google Scholar] [CrossRef]
- Sheiko, S.S.; Vashahi, F.; Morgan, Ḃ.J.; Maw, M.; Dashtimoghadam, E.; Fahimipour, F.; Jacobs, M.; Keith, Ȧ.N.; Vatankhah-Varnosfaderani, M.; Dobrynin, A.V. Mechanically Diverse Gels with Equal Solvent Content. ACS Cent. Sci. 2022, 8, 845–852. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Sheiko, S.S.; Borisov, O.V. Polymer Networks Formed by Molecular Brushes: Scaling Theory. Polym. Sci. Ser. A 2019, 61, 799–804. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Borisov, O.V. Bottlebrush polymer gels: Architectural control over swelling and elastic moduli. Soft Matter 2022, 18, 1239–1246. [Google Scholar] [CrossRef]
- Jacobs, M.; Liang, H.; Dashtimoghadam, E.; Morgan, B.J.; Sheiko, S.S.; Dobrynin, A.V. Nonlinear Elasticity and Swelling of Comb and Bottlebrush Networks. Macromolecules 2019, 52, 5095–5101. [Google Scholar] [CrossRef]
- Kerr, A.; Hartlieb, M.; Sanchis, J.; Smith, T.; Perrier, S. Complex multiblock bottle-brush architectures by RAFT polymerization. Chem.Commun. 2017, 53, 11901–11904. [Google Scholar] [CrossRef]
- Börner, H.G.; Duran, D.; Matyjaszewski, K.; Da Silva, M.; Sheiko, S.S. Synthesis of molecular brushes with gradient in grafting density by atom transfer polymerization. Macromolecules 2002, 35, 3387–3394. [Google Scholar] [CrossRef]
- Liang, X.; Liu, Y.; Huang, J.; Wei, L.; Wang, G. Synthesis and characterization of novel barbwire-like graft polymers poly(ethylene oxide)-g-poly(-caprolactone)4 by the ‘grafting from’ strategy. Polym. Chem. 2015, 6, 466–475. [Google Scholar] [CrossRef]
- Uhrig, D.; Mays, J.W. Synthesis of Combs, Centipedes, and Barbwires: Thinspace Poly(isoprene-graft-styrene) Regular Multigraft Copolymers with Trifunctional, Tetrafunctional, and Hexafunctional Branch Points. Macromolecules 2002, 35, 7182–7190. [Google Scholar] [CrossRef]
- Pelras, T.; Mahon, C.S.; Nonappa; Ikkala, O.; Gröschel, A.H.; Müllner, M. Polymer Nanowires with Highly Precise Internal Morphology and Topography. J. Am. Chem. Soc. 2018, 140, 12736–12740. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.M.; Wilson, C.J.; Wilson, D.A.; Peterca, M.; Imam, M.R.; Perec, V. Dendron-mediated self-assembly, disassembly, and self-organization of complex systems. Chem. Rev. 2009, 109, 6275–6540. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.W.; Ho, D.; Marlow, J.B.; King, J.J.; Hee, C.; Wong, L.N.; Atkin, R.; Smith, N.M.; Warr, G.G.; Norret, M.; et al. Intracellular Communication between Synthetic Macromolecules. JACS 2022, 144, 14112–14120. [Google Scholar] [CrossRef]
- Kröger, M.; Peleg, O.; Halperin, A. From Dendrimers to Dendronized Polymers and Forests: Scaling Theory and its Limitations. Macromolecules 2010, 43, 6213–6224. [Google Scholar] [CrossRef]
- Borisov, O.V.; Polotsky, A.A.; Rud, O.V.; Zhulina, E.B.; Leermakers, F.A.M.; Birshtein, T.M. Dendron Brushes and Dendronized Polymers: A Theoretical Outlook. Soft Matter 2014, 10, 2093–2101. [Google Scholar] [CrossRef]
- Mikhailov, I.V.; Darinskii, A.A.; Zhulina, E.B.; Borisov, O.V.; Leermakers, F.A.M. Persistence length of dendronized polymers: The self-consistent field theory. Soft Matter 2015, 11, 9367–9378. [Google Scholar] [CrossRef]
- Konak, C.; Reschel, T.; Oupicky, D.; Ulbrich, K. Thermally controlled association in aqueous solutions of poly(l-lysine) grafted with poly(N-isopropylacrylamide). Langmuir 2002, 18, 8217–8222. [Google Scholar] [CrossRef]
- Jiang, X.; Lu, G.; Feng, C.; Li, Y.; Huang, X. Poly(acrylic acid)-graft-poly(N-vinylcaprolactam): A novel pH and thermo dual-stimuli responsive system. Polym. Chem. 2013, 4, 3876–3884. [Google Scholar] [CrossRef]
- Zhang, D.; Dashtimoghadam, E.; Fahimipour, F.; Hu, X.; Li, Q.; Bersenev, E.A.; Ivanov, D.A.; Vatankhah-Varnoosfaderani, M.; Sheiko, S.S. Tissue-adaptive materials with independently regulated modulus and transition temperature. Adv. Mater. 2020, 32, 2005314. [Google Scholar] [CrossRef]
- Vashahi, F.; Martinez, M.R.; Dashtimoghadam, E.; Fahimipour, F.; Keith, A.N.; Bersenev, E.A.; Ivanov, D.A.; Zhulina, E.B.; Popryadukhin, P.; Matyjaszewski, K.; et al. Injectable bottlebrush hydrogels with tissue-mimetic mechanical properties. Sci. Adv. 2022, 8, eabm2469. [Google Scholar] [CrossRef]
- Borisov, O.V.; Zhulina, E.B. Amphiphilic Graft Copolymers in a Selective Solvent: Intramolecular Structures and Conformational Transitions. Macromolecules 2005, 38, 2506–2514. [Google Scholar] [CrossRef]
- Kosovan, P.; Kuldova, J.; Limpouchova, Z.; Prochazka, K.; Zhulina, E.B.; Borisov, O.V. Amphiphilic Graft Copolymers in Selective Solvents: Molecular Dynamics Simulations and Scaling Theory. Macromolecules 2009, 42, 6748–6760. [Google Scholar] [CrossRef]
- Prokacheva, V.M.; Rud, O.V.; Uhlik, F.; Borisov, O.V. Intramolecular micellization and nanopatterning in pH-and thermo-responsive molecular brushes. Soft Matter 2020, 16, 208–218. [Google Scholar] [CrossRef]
- Borisov, O.V.; Shavykin, O.V.; Zhulina, E.B. Theory of polyelectrolyte dendrigrafts. Colloid Polym. Sci. 2020, 298, 951–959. [Google Scholar] [CrossRef]
- Birshtein, T.M.; Borisov, O.V.; Zhulina, Y.B.; Khokhlov, A.R.; Yurasova, T.A. Conformations of comb-like macromolecules. Polym. Sci. USSR 1987, 29, 1293–1300. [Google Scholar] [CrossRef]
- Fredrickson, G.H. Surfactant-induced lyotropic behavior of flexible polymer solutions. Macromolecules 1993, 26, 2825–2831. [Google Scholar] [CrossRef]
- Borisov, O.V.; Birshtein, T.M.; Zhulina, Y.B. Temperature-Concentration Diagram of State for Solutions of Comb-like Macromolecules. Polym. Sci. USSR 1987, 29, 1552–1559. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Sheiko, S.S.; Borisov, O.V. Solution and Melts of Barbwire Bottlebrushes: Hierarchical Structure and Scale-Dependent Elasticity. Macromolecules 2019, 52, 1671–1684. [Google Scholar] [CrossRef]
- Sheiko, S.S.; Borisov, O.V.; Prokhorova, S.A.; Möller, M. Cylindrical molecular brushes under poor solvent conditions: Microscopic observations and scaling analysis. Eur. Phys. J. E 2004, 13, 125–131. [Google Scholar] [CrossRef]
- Subbotin, A.V.; Semenov, A.N. Spatial Self-Organization of Comb Macromolecules. Polym. Sci. Ser. A 2007, 49, 1328–1357. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Sheiko, S.S.; Borisov, O.V. Theoretical advances on molecular bottlebrushes: Solutions, gels, and self-assembly. Soft Matter 2022, 18, 8714–8732. [Google Scholar] [CrossRef]
- Feuz, L.; Leermakers, F.A.M.; Textor, M.; Borisov, O. Bending rigidity and induced persistence length of molecular bottle brushes: A self-consistent-field theory. Macromolecules 2005, 38, 8891–8901. [Google Scholar] [CrossRef]
- MSaariaho; Ikkala, O.; Szleifer, I.; Erukhimovich, I.; ten Brinke, G. On lyotropic behavior of molecular bottle-brushes: A Monte Carlo computer simulation study. J. Chem. Phys. 1997, 107, 3267–3276. [Google Scholar] [CrossRef]
- Saariaho, M.; Szleifer, I.; Ikkala, O.; ten Brinke, G. Extended conformations of isolated molecular bottle-brushes: Influence of side-chain topology. Macromol. Theor. Simul. 1998, 7, 211–216. [Google Scholar] [CrossRef]
- Subbotin, A.; Saariaho, M.; Ikkala, O.; ten Brinke, G. Elasticity of comb copolymer cylindrical brushes. Macromolecules 2000, 33, 3447–3452. [Google Scholar] [CrossRef][Green Version]
- Elli, S.; Ganazzoli, F.; Timoshenko, E.G.; Kuznetsov, Y.A.; Connolly, R. Size and persistence length of molecular bottle-brushes by Monte Carlo simulations. J. Chem. Phys. 2004, 120, 6257–6267. [Google Scholar] [CrossRef]
- Theodorakis, P.E.; Hsu, H.-P.; Paul, W.; Binder, K. Computer simulation of bottle-brush polymers with flexible backbone: Good solvent versus theta solvent conditions. J. Chem. Phys. 2011, 135, 164903. [Google Scholar] [CrossRef]
- Cao, Z.; Carrillo, J.-M.Y.; Sheiko, S.S.; Dobrynin, A.V. Computer Simulations of Bottle Brushes: From Melts to Soft Networks. Macromolecules 2015, 48, 5006–5015. [Google Scholar] [CrossRef]
- Hsu, H.-P.; Paul, W.; Binder, K. One- and Two-Component Bottle-Brush Polymers: Simulations Compared toTheoretical Predictions. Macromol. Theory Simul. 2007, 16, 660–689. [Google Scholar] [CrossRef]
- Hsu, H.-P.; Paul, W.; Binder, K. Standard definitions of persistence length do not describe the local intrinsic stiffness of real polymer chains. Macromolecules 2010, 43, 3094–3102. [Google Scholar] [CrossRef]
- Hsu, H.-P.; Paul, W.; Binder, K. Estimation of persistence lengths of semiflexible polymers: Insight from simulations. Polym. Sci. Ser. C 2013, 55, 39–59. [Google Scholar] [CrossRef]
- Muhammadi, E.; Joshi, S.J.; Deshmukh, S.A. Review of computational studies of bottlebrush polymers. Computational Materials Science 2021, 199, 110720. [Google Scholar] [CrossRef]
- Zamurovic, M.; Christodoulou, S.; Vazaios, A.; Iatrou, E.; Pitsikalis, M.; Hadjichristidis, N. Micellization Behavior of Complex Comblike Block Copolymer Architectures. Macromolecules 2007, 40, 5835–5849. [Google Scholar] [CrossRef]
- Li, Z.; Ma, J.; Xheng, C.; Zhang, K.; Wooley, K.L. Synthesis of Hetero-Grafted Amphiphilic Diblock Molecular Brushes and Their Self-Assembly in Aqueous Medium. Macromolecules 2010, 43, 1182–1184. [Google Scholar] [CrossRef]
- Fenyves, R.; Schmutz, M.; Horner, I.J.; Bright, F.V.; Rzaev, J. Aqueous Self-Assembly of Giant Bottlebrush Block Copolymer Sufactants as Shape-Tunable Building Blocks. J. Am. Chem. Soc. 2014, 136, 7762–7770. [Google Scholar] [CrossRef]
- Alaboalirat, M.; Qi, L.; Arrington, K.J.; Qian, S.; Keum, J.K.; Mei, H.; Littrell, K.C.; Sumpter, B.G.; Carrillo, J.-M.Y.; Verduzco, R.; et al. Amphiphilic Bottlebrush Block Copolymers: Analysis of Aqueous Self-Assembly by Small-Angle Neutron Scattering and Surface Tension Measurements. Macromolecules 2019, 52, 465–476. [Google Scholar] [CrossRef]
- Kim, S.; Cho, Y.; Kim, J.H.; Song, S.; Lim, J.; Choi, S.-H.; Char, K. Structural Analysis of Bottlebrush Block Copolymer Micelles Using Small-angle X-ray Scattering. ACS Macro Lett. 2020, 9, 1261–1266. [Google Scholar] [CrossRef]
- Taipaleenmaki, E.; Mouritzen, S.A.; Schattling, P.; Zhang, Y.; Städler, B. Mucopenetrating micelles with a PEG corona. Nanoscale 2017, 9, 18438–18448. [Google Scholar] [CrossRef]
- Wang, Y.; Shao, F.; Sauve, E.R.; Tonge, C.M.; Hudson, Z.M. Self-assembly of giant bottlebrush block copolymer surfactants from luminescent organic electronic materials. Soft Matter 2019, 15, 5421–5430. [Google Scholar] [CrossRef]
- Unsal, H.; Onbulak, S.; Calik, F.; Er-Rafik, M.; Schmutz, M.; Sanyal, A.; Rzayev, J. Interplay between Molecular Packing, Drug Loading, and Core Cross-Linking in Bottlebrush Copolymer Micelles. Macromolecules 2017, 50, 1342–1352. [Google Scholar] [CrossRef]
- Patel, B.B.; Pan, T.; Changc, Y.; Walsha, D.J.; Kwokb, J.J.; Parka, K.S.; Patela, K.; Guironneta, D.; Singa, C.E.; Diao, Y. Concentration-Driven Self-Assembly of PS-b-PLA Bottlebrush Diblock Copolymers in Solution. ACS Polym. Au 2022, 2, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Nguyen, H.V.T.; Johnson, J.A. Mikto-Brush-Arm Star Polymers via Cross-Linking of Dissimilar Bottlebrushes: Synthesis and Solution Morphologies. ACS Macro Lett. 2017, 6, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Henn, D.M.; Holmes, J.A.; Kent, E.W.; Zhao, B. Worm-to-Sphere Shape Transition of Thermoresponsive Linear Molecular Bottlebrushes in Moderately Concentrated Aqueous Solution. J. Phys. Chem. B 2018, 122, 7015–7025. [Google Scholar] [CrossRef] [PubMed]
- Zhulina, E.B.; Borisov, O.V. Micelles Formed by AB Copolymer with Bottlebrush blocks. Scaling Theory. J. Phys. Chem. B 2021, 125, 12603–12616. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, I.O.; Zhulina, E.B.; Borisov, O.V. Self-assembly of bottlebrush block copolymers in selective solvent: Micellar structures. Polymers 2021, 13, 1351. [Google Scholar] [CrossRef]
- De Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA; London, UK, 1979. [Google Scholar]
- Duane, S.; Kennedy, A.D.; Pendleton, B.J.; Roweth, D. Hybrid Monte-Carlo. Phys. Lett. B. 1987, 195, 216–222. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Irback, A. Hybrid Monte Carlo simulation of polymer chains. J. Chem. Phys. 1994, 101, 1661–1667. [Google Scholar] [CrossRef][Green Version]
- Grest, G.S.; Kremer, K. Molecular-Dynamics Simulation for Polymers in the Presence of a Heat Bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef]
- Jones, J.E. On the determination of molecular fields. I. From the variation of the viscosity of a gas with temperature. Proc. R. Soc. London. Ser. A 1924, 106, 441–462. [Google Scholar]
- Behbahani, A.F.; Schneider, L.; Rissanou, A.; Chazirakis, A.; Bacova, P.; Jana, P.K.; Li, W.; Doxastakis, M.; Polinska, P.; Burkhart, C.; et al. Dynamics and Rheology of Polymer Melts via Hierarchical Atomistic, Coarse-Grained, and Slip-Spring Simulations. Macromolecules 2021, 54, 2740–2762. [Google Scholar] [CrossRef]
- McDonald, I.R. NPT-ensemble Monte Carlo calculations for binary liquid mixtures. Mol. Phys. 1971, 23, 41–58. [Google Scholar] [CrossRef]
- Flyvbjerg, H.; Petersen, H.G. Error-Estimates on Averages of Correlated Data. J. Chem. Phys. 1989, 91, 461–466. [Google Scholar] [CrossRef]
- Jin, S.; Collins, L.R. Dynamics of dissolved polymer chains in isotropic turbulence. New J. Phys. 2007, 9, 360. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).