1. Introduction
Hydrogels are three-dimensional (3D) polymeric continuous networks able to entrap a large amount of water (up to 99% of the total weight). They do not dissolve in water, and they maintain their shape due to chemical or physical crosslinking or the entanglement of their polymer chains [
1,
2,
3,
4].
In the particular case of bone tissue engineering (BTE), hydrogels have several advantages: their mechanical properties and degradation can be tuned according to the crosslinking degree; they can provide nutrients because of their ability to swell, incorporating liquids from outside; they can provide an environment suitable for endogenous cell growth. Moreover, hydrogels are absorbable, and they have excellent integration with surrounding biological tissues, limiting the possibility of inflammatory or immune responses [
5].
Hydrogels can be formed by both synthetic and natural derived polymers. Natural polymers have the advantage of being similar to native tissue composition, showing overall better properties compared to their synthetic counterparts, such as low or no cytotoxicity and increased biocompatibility and biodegradability. Among the various natural biopolymers available (i.e., chitosan [
6], keratin [
7], alginate [
8], agarose [
9], cellulose [
10]), silk fibroin (SF) is interesting because of its high mechanical strength, which makes it a good candidate in the case of structural requirements [
11].
SF is the internal protein of silk fiber and is extracted from it as an aqueous solution (regenerated silk fibroin, rSF) by chemical processing [
12,
13] which contributes also to the removal of the immunogenic sericin. SF is biocompatible [
14,
15], bioresorbable [
16,
17], and, when used in the form of gel and film, it is transparent [
18,
19,
20]. Starting with rSF, different procedures have been used to develop hydrogels both by physical and chemical crosslinking.
Physical crosslinking is obtained by inducing the transition of the secondary structure from random coil to β-sheet, which implies the formation of intramolecular hydrogen bonding. Instead, in the chemical crosslinking, the stability is ensured by the formation of covalent bonding and, consequently, a three-dimensional (3D) continuous network [
21]. Chemical crosslinking can be obtained using two approaches. The first consists of promoting the formation of dityrosine and trityrosine crosslinking by the use of different agents with or without an external stimulus (ruthenium and ammonium persulfate and UV irradiation [
22], riboflavin and UVA irradiation [
23], hydrogen peroxide and horseradish peroxidase [
24], genipin in presence of humidity [
25,
26]). The alternative approach is based on the chemical modification of the main SF chain to add reactive moieties that can be activated by external stimuli, like UV light, able to trigger a condensation reaction.
Sil-MA (methacrylated silk) is a chemically modified SF initially designed as ink for 3D fabrication, to be used in digital light processing (DLP) and stereolithography (SLA) 3D printing [
27,
28]. Sil-MA photocrosslinking is achieved by modifying the lysine side groups of the protein, adding a methacrylic functionality, which can generate stable chemical crosslinking bonds upon the addition of a photoinitiator and UV-exposure. Compared to other previously developed silk modification strategies for photocrosslinking [
29,
30,
31], the main advantages is that the entire process is conducted in water, making it attractive for TE applications.
The resulting material is a hydrogel that can be used for 3D fabrication, realizing custom-shaped structures designed by CAD and printed using light-based 3D technologies. This makes the overall technique able to develop patient-specific devices, based on patient X-rays and magnetic resonance imaging, as widely discussed in the literature [
32,
33].
Since its development in 2018, Sil-MA has been widely used as a base material for different architectures, including, but not limited to, bone and cartilage scaffolds [
32,
33,
34,
35,
36], nanoparticles for molecular recognition [
37], and sealant for orthodontic applications [
38]. The use of Sil-MA as an injectable hydrogel has been only recently introduced in the case of spinal cord injury [
39]. The proposed strategy is crosslinking after injection to form the gel in-situ, allowing its stabilization in the injection site [
38,
39]. This has the disadvantage that the possibility of developing complex structures obtainable by 3D-printing is lost, since the whole injected solution is turned into hydrogel, a fact that can also alter the mechanics at the implant site.
In this work, we adapted a previously developed Sil-MA protocol [
27] to a DLP projection and pneumatic extrusion technique that allowed us to print a thin Sil-MA 3D construct capable, once folded, of fully recovering its shape when rehydrated. Sil-MA was produced starting from the raw cocoons and characterized by Fourier Transform Infrared Spectroscopy (FTIR), UV–vis spectroscopy and nuclear magnetic resonance (NMR) to confirm and quantify the methacrylic groups. The rheology of Sil-MA gels and their swelling in water were quantified with different crosslinking times. An initial in vitro biological evaluation using a human lung fibroblast (MRC5) cell line was conducted on the Sil-MA verifying its biocompatibility under the international standard organization standards (ISO 10993). An additional experiment was conducted using human-adipose-derived stem cells (ADSCs) to prove if Sil-MA can support osteogenesis. Finally, we developed a prototypical grid structure based on Sil-MA to perform a withdraw and release assay, demonstrating the shape recovery ability after the ejection from an insertion needle and following re-hydration.
3. Discussion
Sil-MA is a methacrylated biopolymer developed to fabricate SF constructs using light-based 3D additive manufacturing. In this work, we developed a prototypical grid structure based on Sil-MA, which showed three main characteristics: it can be folded into a small volume, can be ejected through an insertion needle, and can fully recover its shape after complete rehydration.
The material characterization has revealed that we were able to successfully methacrylate the SF with a rate of functionalization of about 30% (demonstrated by TNBs assay and NMR), which is similar to the rate reported in previous works for the same reaction [
27,
34].
Sil-MA-based hydrogels have been shown in the literature to have good mechanical performances, increasing the interest in the TE applications. In particular, Sil-MA gels were reported having compressive and elastic moduli increasing with Sil-MA concentration, achieving 130 kPA in compression and 15 kPa in tension for gels produced with a 30% Sil-MA solution [
27]; these values are close to the elasticity modulus of the normal bone extracellular matrix (ECM), 20–50 kPa [
42], and far from the elasticity modulus of the mineralized bone, whose values are as large as 10–20 GPa [
43]. Herein, we did not test the Sil-MA constructs by compression given their thickness. Therefore, this construct is not suitable for load-bearing applications but rather for cavities or surface filling.
Other silk-related materials were developed with the specific purpose of structural applications [
11,
25]. The rheological test performed gave results of G′ and G″ higher than those reported in literature [
39]; this is easily understandable, considering that, even though the starting material was the same, the crosslinking degree after UV-exposure depends on a series of other factors, which vary from experiment to experiment (adsorbed energy, wavelength of the UV lamp, concentration of Sil-MA, and amount of LAP).
Sil-MA has been widely used as bioink and proven to be non-cytotoxic when cells were included in the ink [
27,
38]. The vitality was reported to be UV-exposure-dependent and to also have a dependency on the amount of added photoinitiator that, above a certain amount, results in cytotoxicity [
34]. In addition, when cells were included into the material to perform 3D cell cultures, the increase in the Sil-MA concentration decreased the cell viability [
39]. The threshold for the 3D cell culture resulted in a Sil-MA concentration of 10% [
39]. In this study, Sil-MA has not been used as bioink, which, by definition, requires cells prior to bioprinting, but as a base material to build implantable construct. For this reason we could perform the 3D printing with a higher Sil-MA concentration (up to 20%) without compromising the cells vitality. The material was tested, for the first time to our knowledge, following the international standards organization protocols (ISO-10993) on the MRC5 cell line to certify its biocompatibility and indicated an absence of cytotoxicity. Moreover, additional in vitro biological evaluation on MRC5 cells lines showed good adhesion, proliferation, and metabolic activity. However, MRC5 in the first days showed low proliferation compared to rSF, which could be partially explained by the possible residual presence of GMA or LiBr, which inhibit the proliferation ability of cells. Previous authors showed that the ideal dialysis period for the modification of SF and GMA is seven days to preserve a good cell proliferation rate [
44].
We also performed a test to evaluate the osteoinductivity of Sil-MA using human ADSCs to prove if Sil-MA can support osteogenesis. Our data confirmed that methacrylation does not negatively interfere with osteogenesis and that Sil-MA displays osteoconductive properties similar to those of SF sponges in our previous study [
45].
In terms of 3D printing, the DLP projector ensured better-resolved features when compared with pneumatic extrusion. This was expected, considering the higher resolution and precision of the optical system. However, if compared to the literature [
27], our DLP-printed structures did not reach the theoretical resolution of the projector. This can be be attributed to the manual focusing and the consequent broadening of the structure when compared to the design. When compared to GelMA printed with a DLP, the resolution obtainable is similar. In fact, GelMA has been recently printed with a resolution of 200 μm by a DLP printer with a maximum resolution of 50 μm [
46]. When pneumatic extrusion was used, an increase in the dimension occurred in the Sil-MA structures and should be attributed to the flow stresses relaxation, a phenomenon widely discussed in the literature [
40,
41].
Other authors previously showed memory-shape features of 3D-printed scaffolds made of fibroin. As an example, a memory-shape-implant meniscus was produced by an enzymatic cross-link of rSF with horseradish peroxidase [
47], but in this case, the produced structure was not injectable. An injectable, photoluminescent, carbon-nanotubes-doped sericin scaffold (CNTs-SS) with a programmable shape-memory property was developed for stroke applications, showing how this type of scaffolds can be readily compressed into small volumetric mass (roughly 90%) and conveniently injected into stroke cavities, followed by shape restoration triggered by fluids [
48]. However, to our knowledge the possibility of injecting a SF-structured scaffold was not reported before.
While promising, our study has several limitations. First, we failed to perform multilayer structures in case of DLP because of the loss of resolution; further studies need to be performed to tune this aspect. In case of pneumatic extrusion, we reached the printing of 10 layers, thanks to the poloxamer support bath, but also in this case with an important loss in terms of resolution.
Further studies should be performed on the 3D-printed constructs to evaluate cell seeding, adhesion, proliferation, and differentiation in vitro, as well as neo tissue formation and biodegradability in vivo. Whether these shape-memory properties would be maintained in vivo has yet to be confirmed: further in vivo analysis should be performed to successfully prove the possibility of inserting the scaffold into a bone cavity or surface and verify the effectiveness in recovering its shape to fit the whole space and the capacity to promote bone formation.
5. Materials and Methods
5.1. Sil-MA Preparation
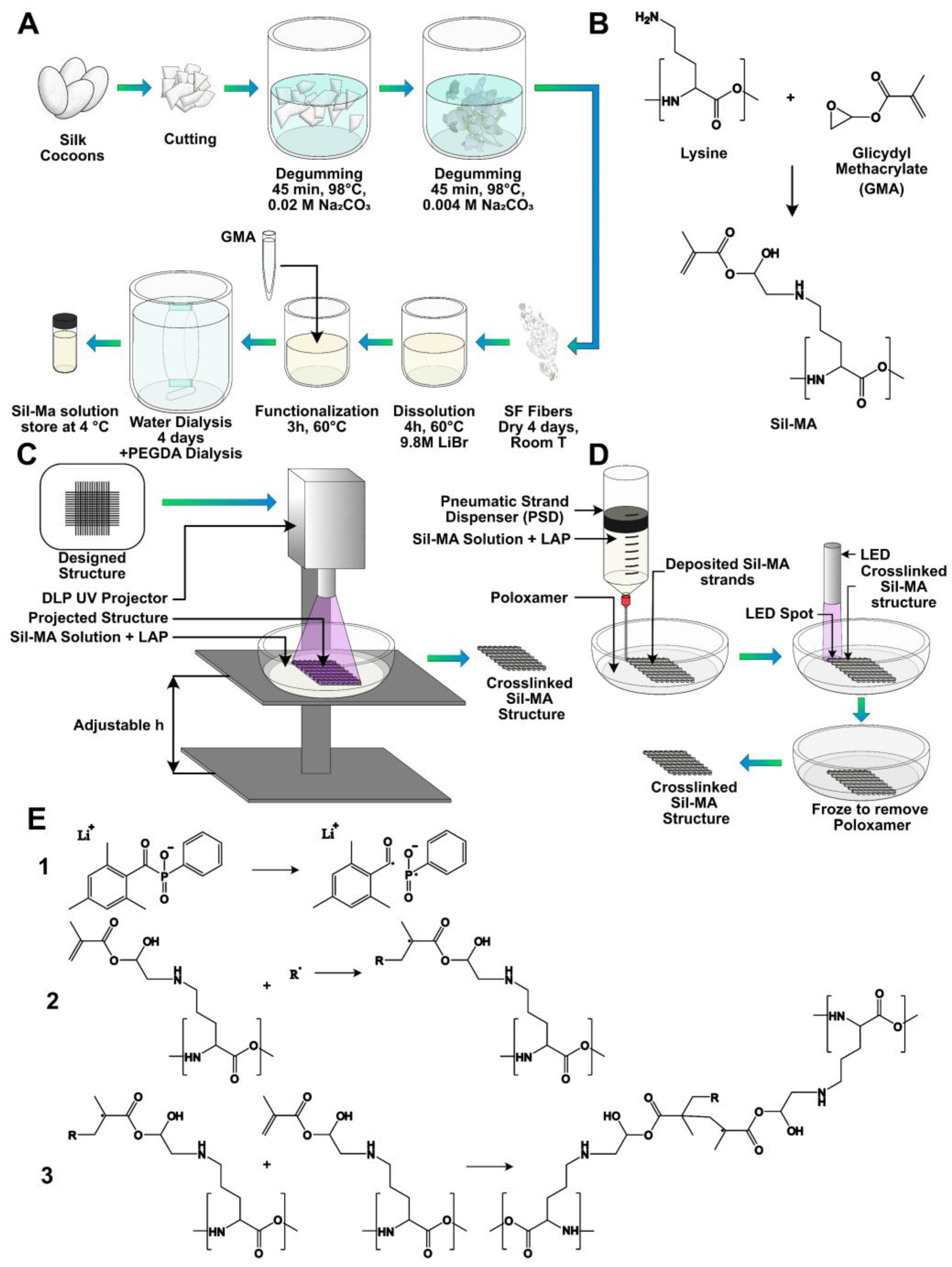
Bombyx mori silkworm cocoons were obtained from Chul Thai Silk Co., Phetchaban, Thailand, and degummed for extracting SF, following a previously published protocol briefly illustrated in
Figure 7A [
12]. Each silkworm cocoon was cut into two pieces and delaminated into two foils. A unit of 30 g of delaminated cocoon was first boiled for 45 min at 98 °C in a boiling bath containing an aqueous solution of 0.02 M Na
2CO
3 (3.3 g of sodium carbonate, Sigma-Aldrich, in 3 l of distilled water) and then boiled for other 45 min at 98 °C in a boiling bath containing an aqueous solution of 0.004 M Na
2CO
3 (1.2 g of sodium carbonate in 3 l of distilled water). For completely removing sericin and the residual salt, SF was rinsed out in distilled water, lowering the bath temperature step by step, in order to not expose SF to high temperature gradients. When room temperature was reached, SF was further washed in distilled water. The degummed SF was dried under the hood at room temperature for 48 h. Degummed SF was dissolved in a 9.3 M LiBr (Honeywell Fluka, Cat. 746479) previously prepared, at 20%
w/
v concentration for 3 h at 65 °C in the oven. SF must be completely dissolved and appear clear after 3 h.
Successively, glycidyl methacrylate (GMA, Sigma-Aldrich, 151238) was added in the beaker in this proportion: 1 mL of GMA per 4 g of SF, chosen according to Soon Hee Kim et al. [
27]. The reaction was carried out, stirring with 300 rpm at 60 °C for 3 h. The hypothesized chemical reaction of the modification is reported in
Figure 7B: a di-β-hydroxyamide group is supposed to form from the primary amine (of the lysine side group) in SF and the epoxy ring of GMA, through the nucleophilic substitution of nitrogen on one carbon atom of the epoxy ring and the consequent ring opening. After reaction, the solution appeared cloudy. The mixture obtained was dialyzed against distilled water using a 3.5 kDa cutoff dialysis membrane for 4 days, in order to remove excess salt and GMA, which is a toxic reagent. At the end, the concentration was measured by UV spectroscopy (Nanordop 1000, ThermoFisher Scientific, Waltham, MA, USA), and the solution was dialyzed again against a 25%
w/
v concentrated PEGDA solution, using a 3.5 kDa cutoff dialysis cassette to concentrate Sil-MA. The dialysis was stopped when the solution concentration of Sil-MA reached 10%
w/
v.
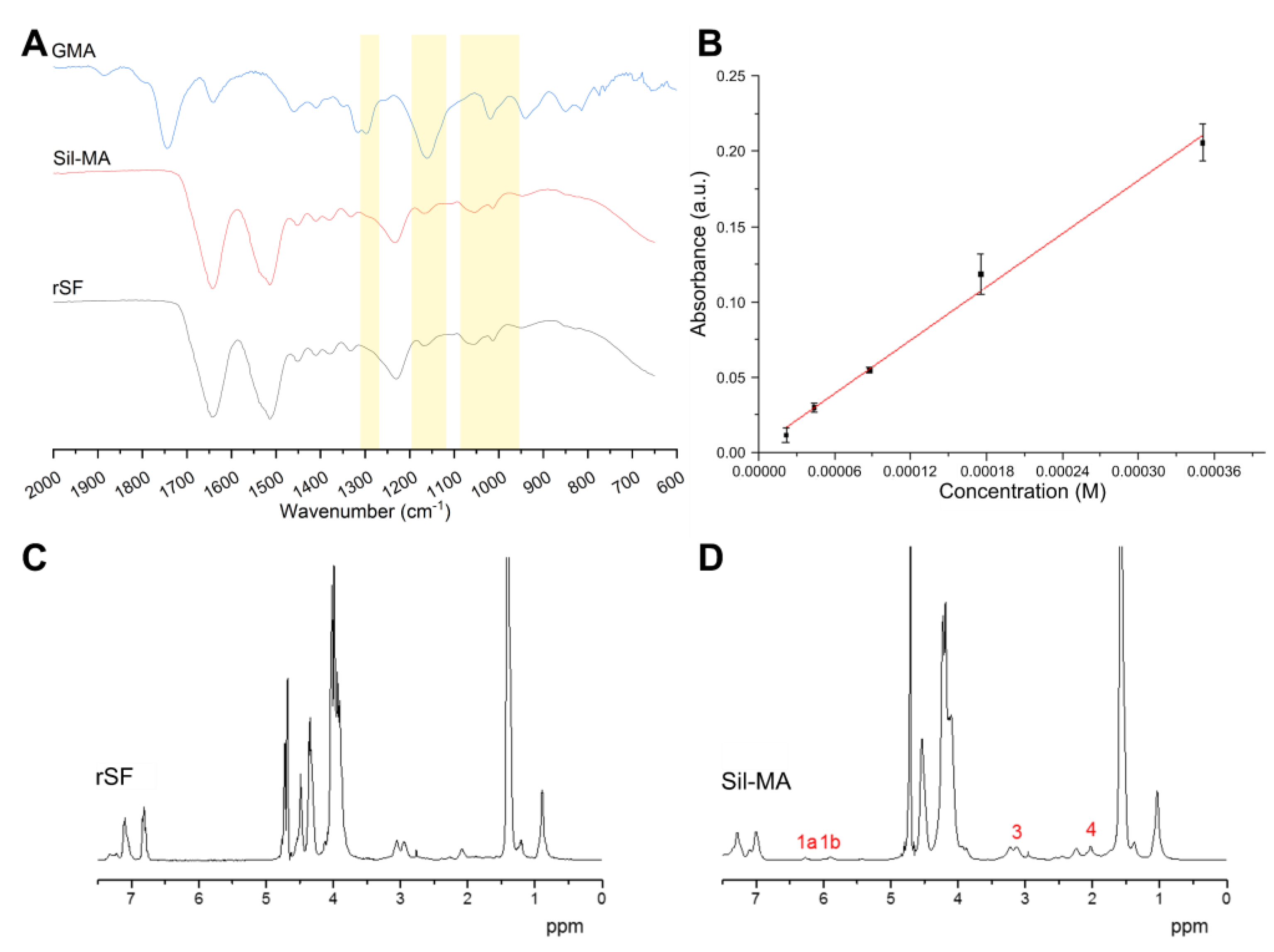
5.2. Infrared Spetroscopy
FTIR spectroscopy was used to qualitatively confirm the presence of methacrylic groups in the Sil-MA structure. A FTIR spectrometer (Spectrum ONE, PerkinElmer, Waltham, MA, USA) was used, and the samples were analyzed in UATR (universal attenuated total reflectance) mode. The wavenumber range analyzed was 4000–650 cm−1. The spectra were acquired as a mean of 16 scans with a resolution of 1 cm−1.
5.3. UV Specrtoscopy
A quantitative analysis was done by means of a 2,4,6-trinitrobenzene sulfonic acid (TNBS) assay, a sensitive assay reagent for the determination of free amino groups, in order to quantify the methacrylation degree in Sil-MATNBS; upon reaction with primary amines, it forms a highly chromogenic product whose absorbance at 335 nm to 345 nm can be measured. As GMA reacts with free amino groups in SF, which are mostly present in lysine, the calculation of the remaining free amino groups in Sil-MA can be used to identify the methacrylation degree. TNBS assay was performed on SF and Sil-MA to quantify the difference in free amino groups.
TNBS (Sigma-Aldrich, 92822) was diluted in a 0.1 M sodium bicarbonate buffer (pH = 8.5). The stock solutions for the samples were prepared at a concentration of 2.43 mg/mL, measured by NanoDrop. Then, 100 µL of each sample stock solutions were incubated at 37 °C for 2 h in 100 µL of TNBS solution. To stop and stabilize the reaction, 250 µL of 10%
w/
v SDS and 125 µL of 1 M HCl were added to each sample after incubation. The calibration curve was generated by the use of several standard samples composed of water solutions containing increasing concentrations of beta alanine (PHR1349, Sigma Aldrich, St. Louis, MO, USA), which is known to have one amino group. The used concentrations are reported in
Table 3. All of the absorbance measurements were taken using a microplate reader (Spark 10M, Tecan, Hombrechtikon, Switzerland), at 340 nm. All of the absorbance measurements were subtracted from the value of the blank (consisting of all the reagents used, without beta alanine).
The analysis is based on the Lambert–Beer law (Equation (2)). The calibration curve can be generally represented by Equation (3), and, by inverting it (Equation (4)), we were able to calculate the concentration of aminoacidic groups in the bare SF (C
rSF), the concentration of the aminoacidic group in the modified Sil-MA (C
Sil-MA), and the percentage of substitution (DS, Equation (5)).
5.4. Nuclear Magnetic Resonance (NMR)
In order to confirm the result of quantitative optical spectroscopy, SF and sil-MA were examined by 1H liquid NMR using a Bruker (Billerica, MA, USA) Ultrashield Plus spectrometer (9.4 T) at a frequency of 400 MHz. A total of 100 mg of each sample was dissolved in 500 µL of a 9.3 M LiBr solution. Then they were diluted in D
2O. The degree of methacrylation was defined according to the lysine groups in SF that are modified in Sil-MA. The spectra were analyzed removing the signal of deuterium, and the signal at 1 ppm was used to normalize each spectrum. Then the lysine signal (2.9 ppm) of the two samples was integrated. The substitution degree was calculated following Equation (6).
5.5. Water Uptake
A total of 500 µL of Sil-MA were cured following the crosslinking procedure explained above. Two curing times were considered: 5 s and 30 s. Furthermore, 5 mL vials made of polystyrene transparent to UV light were used. The samples were weighed just after crosslinking to obtain
Wcross. Then they were hydrated for 0.5, 1, 2, 4, 7, 24, 48, and 72 h in distilled water and maintained in a refrigerator during the test. Water was removed at each sampling time, and hydrogels were weighed to obtain
Wswallen. The water uptake was calculated according to Equation (7).
5.6. Rheology
Rheological measurements were performed to understand how the mechanical properties change with changing curing time. The rheological properties of Sil-MA hydrogels were measured using a stress control rheometer (Discovery HR-2, TA Instruments) equipped with a Peltier plate for temperature control. The measurements were performed using parallel plate geometry (40 mm in diameter) at 25 °C. Disc-shaped samples 40 mm in diameter and 2 mm thickness were crosslinked using a cylindrical mold and a UV LED lamp. Three samples with different curing times were analyzed: 5 s and 30 s. The storage modulus (G′) and loss modulus (G″) were measured in oscillatory mode. Firstly, all of the samples underwent a strain sweep test in order to find the linear viscoelastic region, then frequency sweep tests were performed inside this region. In the strain sweep measurements, the samples were tested, keeping constant angular frequency at 10 rad/s and varying the strain from 0.1 % to 30%, acquiring five points per decade in logarithmic scale. For frequency sweep measurements, the samples were tested at varying angular frequency from 0.1 to 25 rad/s (logarithmic scale, 5 points pe decade), keeping shear strain constant at 1%. One sample cured for 5 s was also tested from 0.1 to 100 rad/s. All of the tests were carried out under axial force control (2 N) during gap closure.
5.7. Morphology
The morphology of the construct was evaluated by optical microscopy (Eclipse 90i, Nikon, USA) comparing the printed object with the design to evaluate its consistency. The images were analyzed by FIJI (v. 1.53t, National Institute of Health, Bethesda, MD, USA).
5.8. In-Vitro Biological Evaluation
Sil-MA and rSF were tested in the form of film, both prepared with the same methodology. The lyophilized rSF was dissolved in formic acid at 8% w/v concentration. Sil-MA films were produced starting from a sil-MA water solution with a 10% w/v concentration and adding 0.1% w/v of LAP photoinitiator. The obtained mixtures were poured and cast in a Petri dish overnight. When all of the formic acid was evaporated, rSF and Sil-MA films were exposed to UV light for 30 s to stabilize them. All films were washed twice in distilled water to eliminate possible formic acid residues. rSF and Sil-MA were sterilized using 70% ethanol for 30 min at room temperature before being placed on 48-well plates.
Biological tests were performed on Sil-MA and rSF to verify the potential use of this material in TE. A control of cells cultured in adhesion was also used (ctrl−, TCP). The effects of Sil-MA were evaluated on cytotoxicity, cell metabolism, and cell adhesion using the human lung fibroblast (MRC5) cell line, in compliance with the European Standard EN ISO 10993-12:2004 and 10993-5:2009.
MRC5 were expanded upon reaching 70% confluence. Then they were detached and seeded at 15 × 103 cells/cm2 into 48-well plates containing Sil-MA substrates.
5.8.1. Lactate Dehydrogenase (LDH) Assay
Cytotoxicity was evaluated by means of an LDH assay (ThermoFisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions. Four experimental groups were tested, as described in
Table 4. The MRC5 cell line as seeded in a 96-well tissue culture plate at a concentration of 5000 cells/well and cultured in standard medium until about 70% confluence. Then the medium was replaced with 100 µL/well of phenol red and with heat-inactivated serum. The volume of the medium was calculated according to ISO-10993-12:2004 (small, molded items, thickness: 0.5–1 mm, extraction ratio area/volume: 3 cm
2/mL). The surnatant was harvested at 48 h of culture and analyzed. For each group, five technical replicates were performed. The colorimetric detection of LDH was performed at 492–620 nm on a spectrophotometer, and cytotoxicity was calculated as a fold decrease compared to the positive control (100% of cytotoxicity).
5.8.2. Alamar Blue Assay
Metabolic activity was evaluated by means of an Alamar Blue assay. MRC5 cells line were seeded on the different experimental groups (Sil-MA films, rSF films, on adhesion-TCP). After 1, 3, and 6 days of culture, the medium was discarded, and the samples were incubated with 1 mL of medium containing 10% Alamar Blue reagent (Resazurin, R7017, Sigma Aldrich, St. Louis, MO, USA) for 1.5 h at 37 °C. Then, 100 µL of the supernatant were transferred to a black 96-well plate; technical duplicates were performed for each sample. A negative control, 100 µL of medium containing 10% Alamar Blue reagent, was also analyzed. The fluorescence intensity was measured using a microplate reader (Spark 10M, Tecan): the excitation wavelength was 590 nm, while the emission wavelength was 535 nm.
After the analysis, the seeded films were washed twice with PBS and successively used to perform the Pico Green assay.
5.8.3. Pico Green Assay
Cell adhesion and proliferation was evaluated by using a Pico Green fluorescence assay kit (Pico Green assay, P11496, Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions, which quantifies the DNA in biological samples and thus the cell number. At each time point (day 1, day 3, day 6), 250 µL of 0.05% Triton-X were poured in each well, and after 20 min, the well plate was moved to a −20 °C refrigerator until the test was carried out. The procedure needed a calibration step, in which samples with known DNA concentration were used in order to build a calibration curve. A solution of the Tris-EDTA buffer was prepared, diluting the Tris-EDTA buffer 20 times provided by the kit with distilled water. A DNA working solution was prepared by adding 20 µL of the stock solution (100 µg/mL) in 980 µL of the Tris-EDTA buffer. The samples for the standard curve were prepared using different composition of the Tris-EDTA buffer and DNA working solution (
Table 5).
Each sample was sonicated for 10 s and diluted 1:1 with the Tris-EDTA buffer to obtain 500 µL of final volume. The Pico Green working solution was prepared by diluting 50 µL of Pico Green stock solution in 10 mL of Tris-EDTA buffer (200-fold dilution). A total of 100 µL of all the samples was moved to a black 96-well plate in triplicates. A total of 100 µL of the Pico Green working solution as added to each well and incubated at room temperature for 2–3 min. The fluorescence intensity was measured using a microplate reader (Spark 10M, Tecan): the excitation wavelength was 485 nm, while the emission wavelength was 535 nm.
5.9. Confocal Microscopy
Confocal analysis (Nikon A1 Confocal Microscope, ) was performed in order to estimate cell adhesion on Sil-MA films, compared to rSF and cells cultured in adhesion. At day 1, 3, and 6, cells were fixed with 4% paraformaldehyde (PFA) at room T for 20 min. Successively, cell membrane was permeabilized using 0.2 % Triton X-100 solution, and nuclei were stained with DAPI (5.4 µL in 25 mL PBS), whereas the cytoskeleton actin was stained with Phalloidin i-Fluor 488 (5 µL in 25 mL PBS) (ab176753, Abcam, Cambridge, UK). After staining, the samples were washed three times with PBS and stored at 4 °C.
5.10. Osteogenesis
To investigate whether the constructs could play a role in supporting osteogenic differentiation, human ADSCs (Lonza, Basel, Switzerland) were seeded onto the Sil-MA films and rSF films or in adhesion and cultured under osteogenic condition for 14 days in α-MEM 20% FBS supplemented with 100 nM dexamethasone, 100 µM ascorbic acid, and 10 mM β-glycerophosphate. At Day 14, Alizarin Red S (AR-S) (Sigma Aldrich, St. Louis, MO, USA) staining was performed to assess the presence and extent of mineralization. Briefly, cells were stained with 40 mM AR-S for 20 min after being fixed for 15 min at RT in formaldehyde (Kaltek, Padova, Italy) in 10% phosphate buffered saline (PBS) and washed twice with PBS. A spectrophotometric analysis with a TECAN Infinite® 200 PRO (Tecan Italia S.r.l., Cernusco Sul Naviglio, Italy) was performed to quantify the mineral apposition.
5.11. UV Crosslinking
A water solution of LAP photoinitiator at 1%
w/
v concentration was prepared. LAP was added to the Sil-MA solution at 0.1%
w/
v concentration. The obtained mixture was exposed to 365 nm UV light using a UV LED lamp (Spot LED 15 W, Unionprint, Milan, Italy). The crosslinking proposed mechanism is shown in
Figure 7E. The lamp was placed at a distance of 5 cm from the support plane. In this position, an irradiated spot with a 7 cm diameter was considered. The intensity at 5 cm from the lamp was about 433 W/m
2 (it was measured with D0971 quantum photo-radiometer and thermometer data-logger by Delta Ohm using the probe for wavelengths of 315–400 nm).
5.12. DLP Printing
Structures were developed using a digital light processing (DLP) UV projector (PRO4500 DLP, Wintech, San Marcos, CA, USA) mounted on a custom-made setup. The set-up used is shown in
Figure 7C. The sample was poured in a Petri dish fixed to the support plane, and the focus was regulated by means of a screw moving the support plane along the vertical axis. The intensity of the light at 4 cm from the source was about 45 W/m
2 (measured with a D0971 quantum photo-radiometer and a thermometer data-logger by Delta Ohm, using the probe for wavelengths of 315–400 nm). The emission peak of the projector LED was at 370 nm, and the resolution of the projected image was 1140 × 912 pixels.
After adding LAP to the Sil-MA solution at 0.1% w/v concentration, 500 µL of the obtained mixture was poured into a Petri dish with 30 mm in diameter and exposed to UV light for 60 s. Patterns were developed by pipetting distilled water on the sample, which allowed uncrosslinked fibroin removal.
Some trials in fabricating 3D constructs were done using a layer-by-layer approach: after a first layer was crosslinked, the procedure was repeated, adding another 500 µL of solution in the Petri dish and lowering the support plane in order to adjust the focus. The time of UV exposure was 60 s for each layer. The development was done after the crosslinking of the last layer.
5.13. Pneumatic Extrusion Printing
The 3D-printing process was also performed by means of a 3D Discovery platform (RegenHU, Villaz-St-Pierre, Switzerland). The procedure is briefly schematized in
Figure 7D. First, the structures were designed by means of dedicated CAD software (BioCAD, RegenHU, Villaz-St-Pierre, Switzerland). The scaffolds were realized with a 10 × 10 mm square base and a total height of 2 mm. A 0/90° infill pattern was selected as the internal microarchitecture. The fiber diameter was set to 200 µm and pore size to 800 µm. A layer height of 200 µm was chosen, leading to the stacking of 10 layers to achieve the desired construct height. The design software enabled us to set the printing process to be automatically replicated within six-well plates. The wells were filled with a support gel to retain the ink fibers’ shape until crosslinking was applied, so as to improve the fabricated constructs’ fidelity to the design.
The support gel was formulated by dissolving Poloxamer 407 (Sigma-Aldrich St Louis, MO, USA) in distilled water, in a 30% w/v ratio. The gel was formed by gradually adding the polymer to the solvent under magnetic stirring at a temperature of 4 °C to facilitate dissolution. Subsequently, the plate wells were filled with the gel and kept at RT for a minimum of 30 min, until gelation was observed, prior to the printing process.
To fabricate the 3D constructs, the Sil-MA cartridge was loaded in a pneumatic extrusion printhead of the 3D discovery platform. A 200 µm nozzle with a length of 12.7 mm was used. The process was performed at room temperature (RT), with a pressure of 0.6 bar and a printing speed of 10 mm/s. After fabrication, each scaffold was irradiated with a 365 nm UV lamp available in the 3D discovery platform. The photocrosslinking duration was set to 5 min.
Finally, the six-well plates were kept at 4 °C for a time of at least 30 min, until sol transition was observed. The 3D scaffolds were then removed from the wells and rinsed with PBS three times (5 min per cycle) to remove poloxamer support gel residuals.
5.14. Characterization of Shape-Memory Property
A proof of principle of the shape-memory property was performed by the immersion of the sil-MA construct in PBS for 30 min. Afterwards, the structure was compressed by aspiration in a pipette cap with a diameter of 2 mm. Then, the construct was extruded from the cap and rehydrated by the addition of PBS until achieving complete shape recovery.
5.15. Statitical Tests
The statistical tests were conducted using R (v. 4.1.3, AT&T, Dallas, TX, USA) in R studio (v. 1.4.1106, Posit, Boston, MA, USA) as IDE. An ANOVA test with a post-hoc Tukey test was performed to verify if there was a statistical difference among the groups. Groups were considered statistically different with a p < 0.05. The confidence levels were assigned as follows: p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***).


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}