Influence of Ceria Addition on Crystallization Behavior and Properties of Mesoporous Bioactive Glasses in the SiO2–CaO–P2O5–CeO2 System

 ,
,  ,
,
Abstract
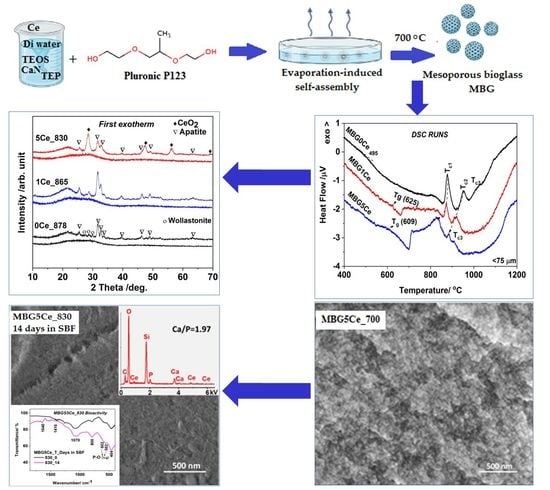
:
1. Introduction
2. Results and Discussion
2.1. Phase Evaluation in the G(0/1/5)Ce Gels
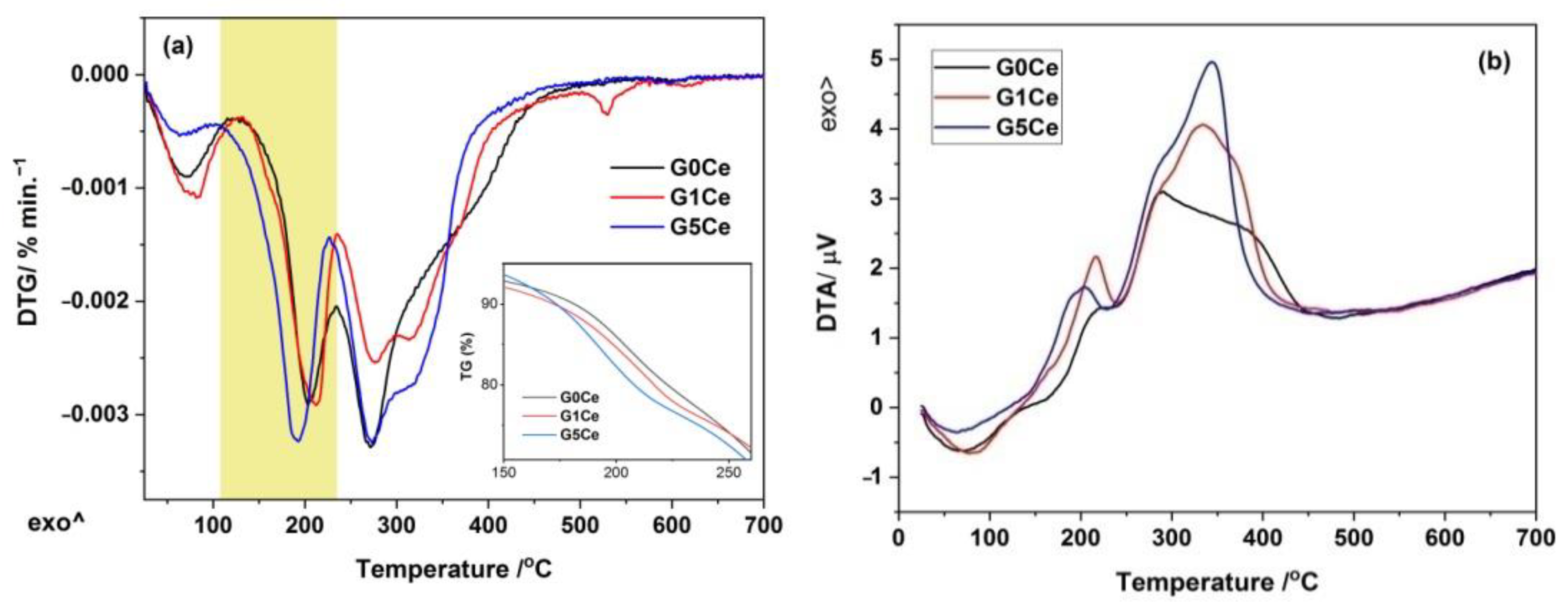
2.2. DTA/DTG/TG Analysis of the G(0/1/5)Ce Gels
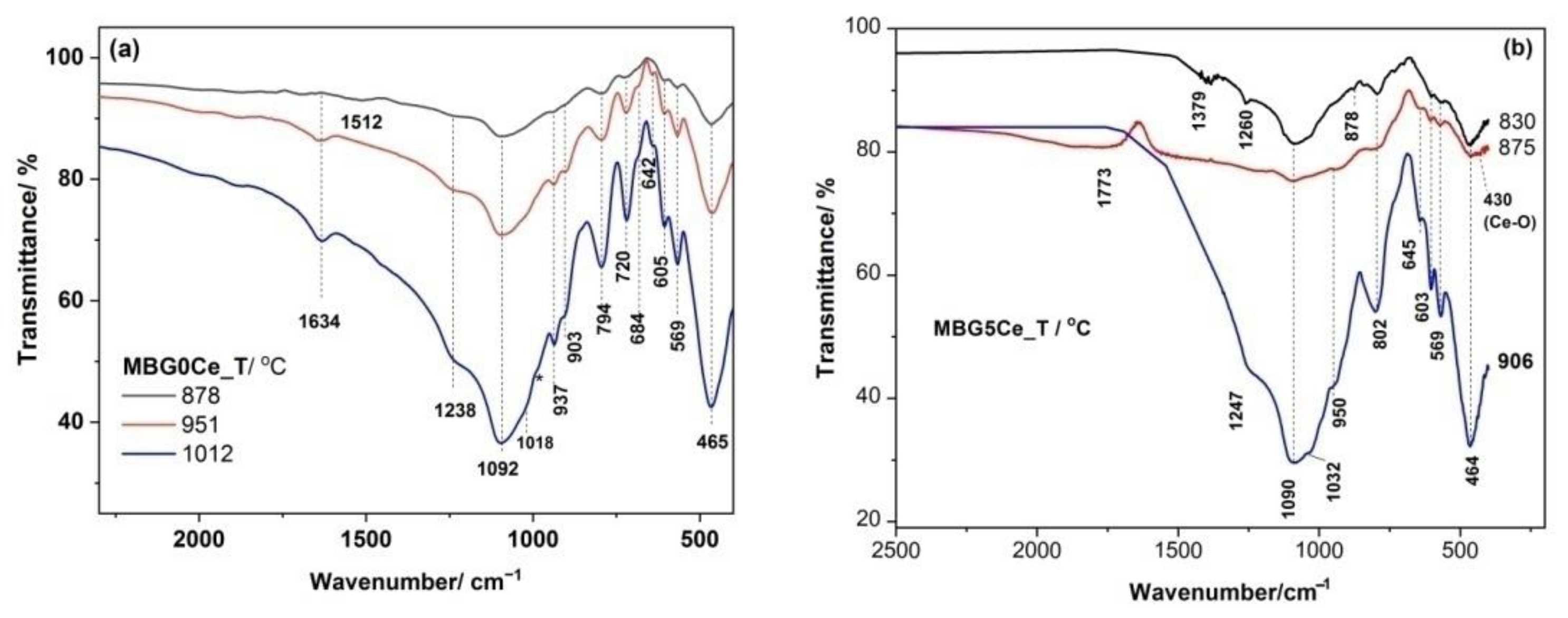
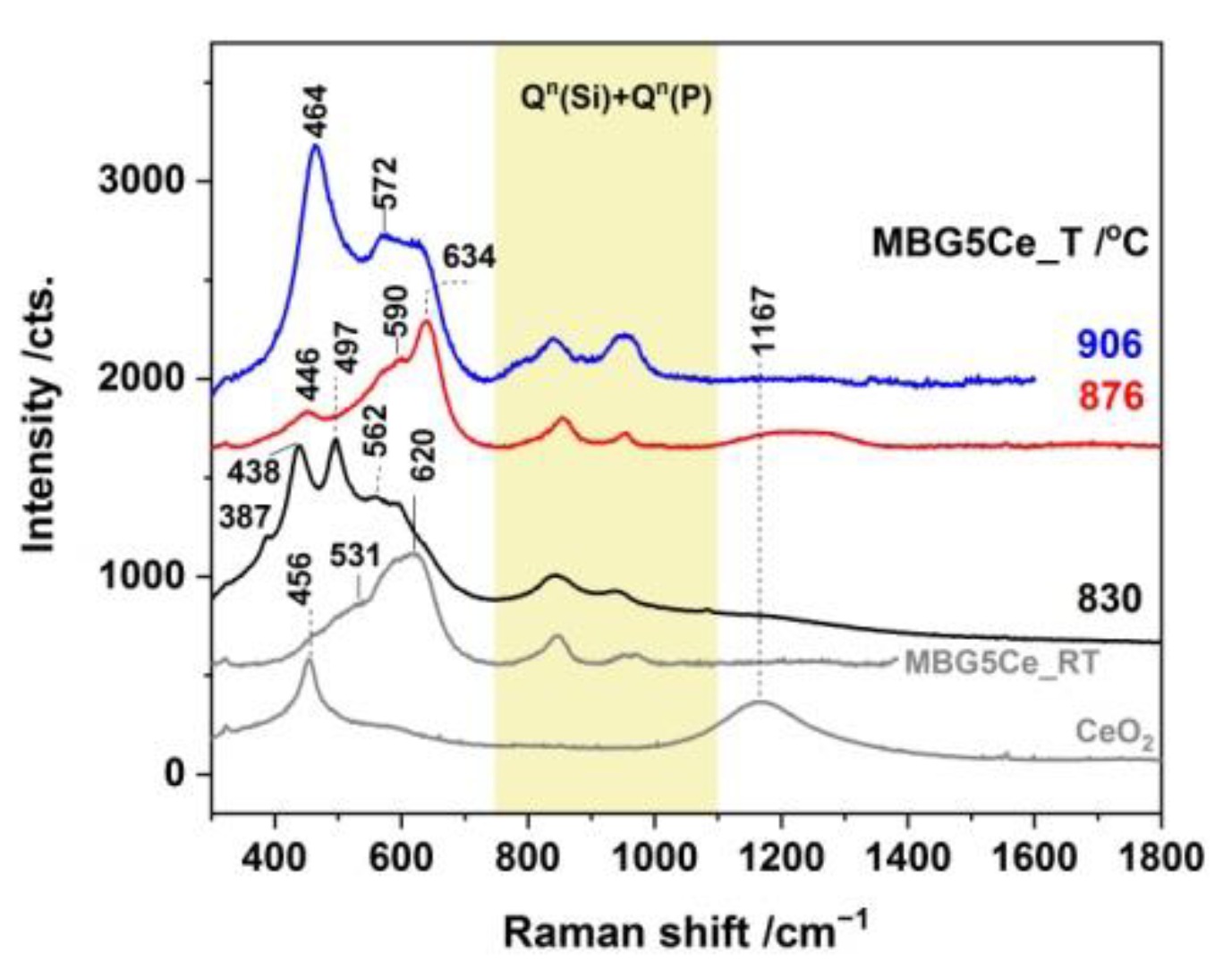
2.3. Phase Identification in the Devitrified Ce-Containing MBGs
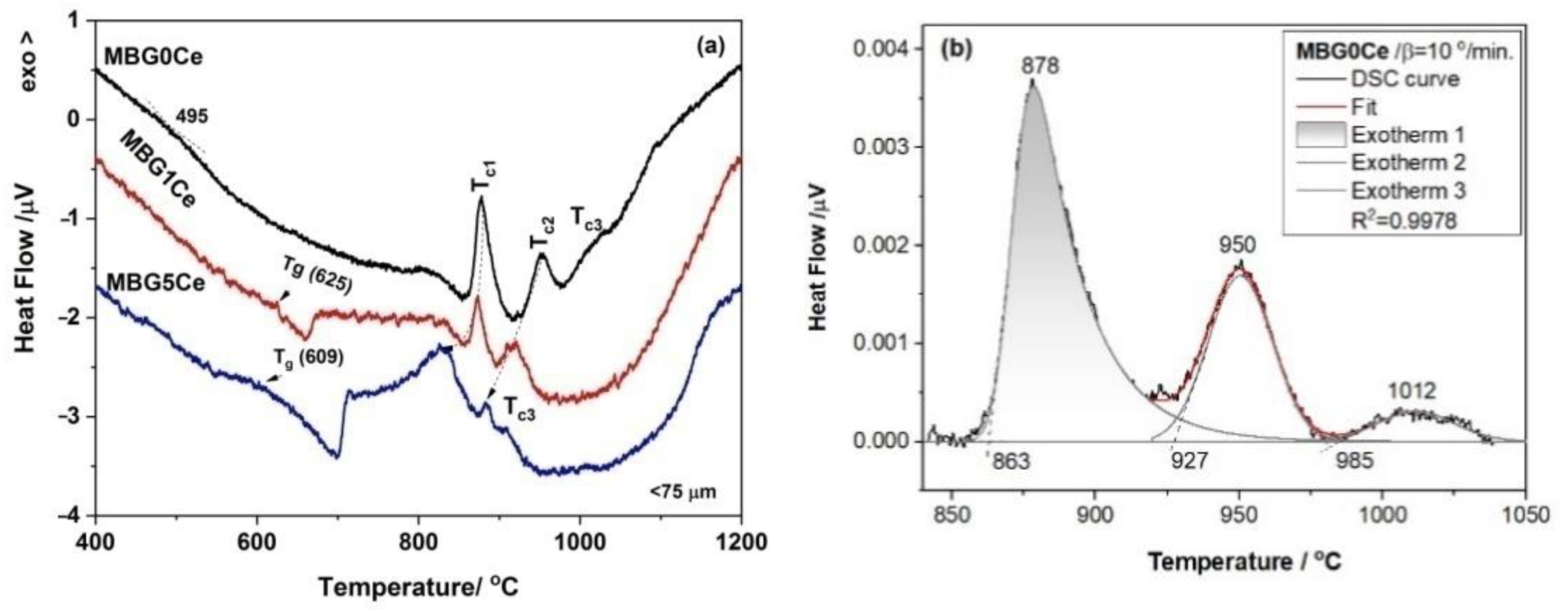
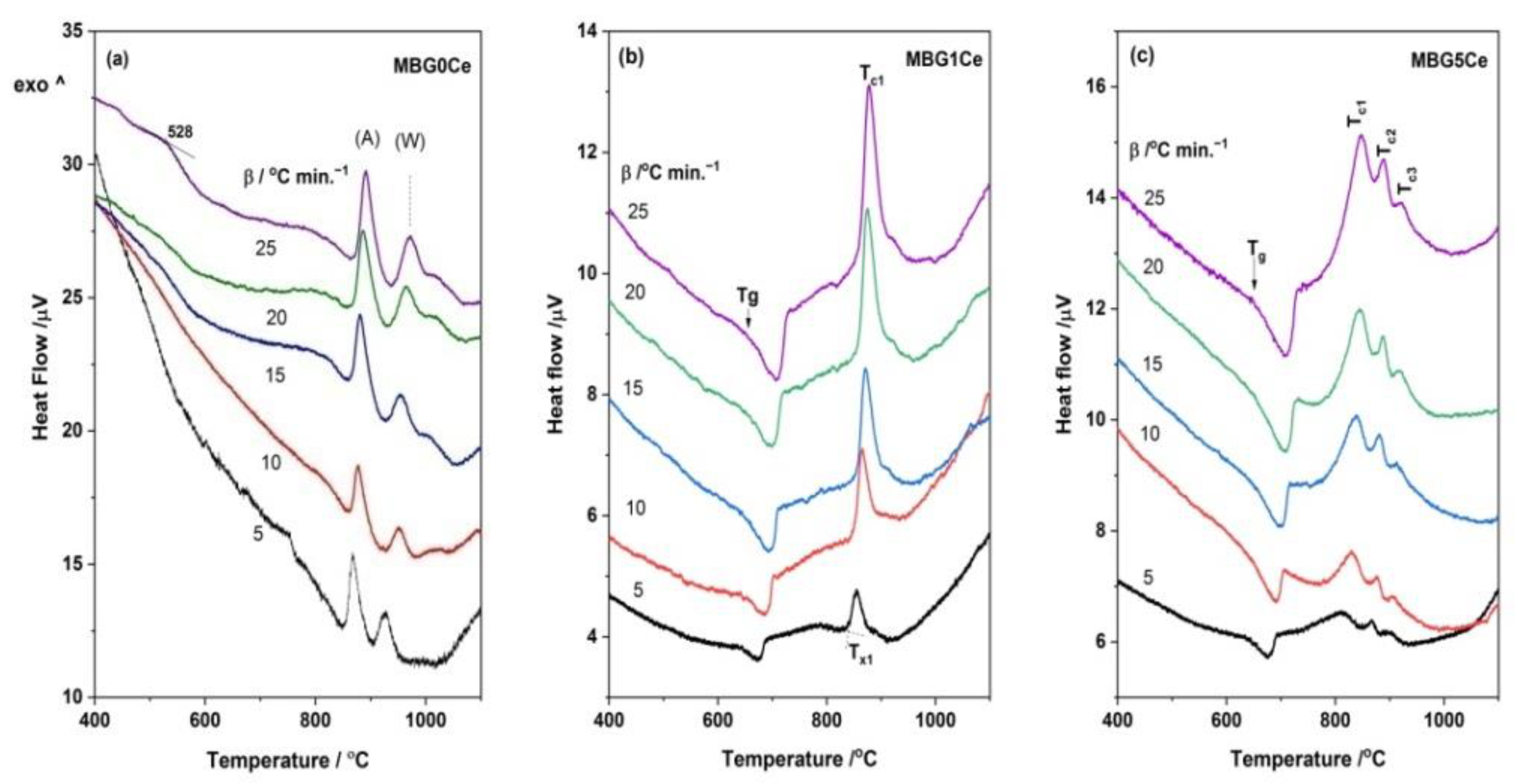
2.4. Crystallization Behavior of the Stabilized Ce-Containing MBGs
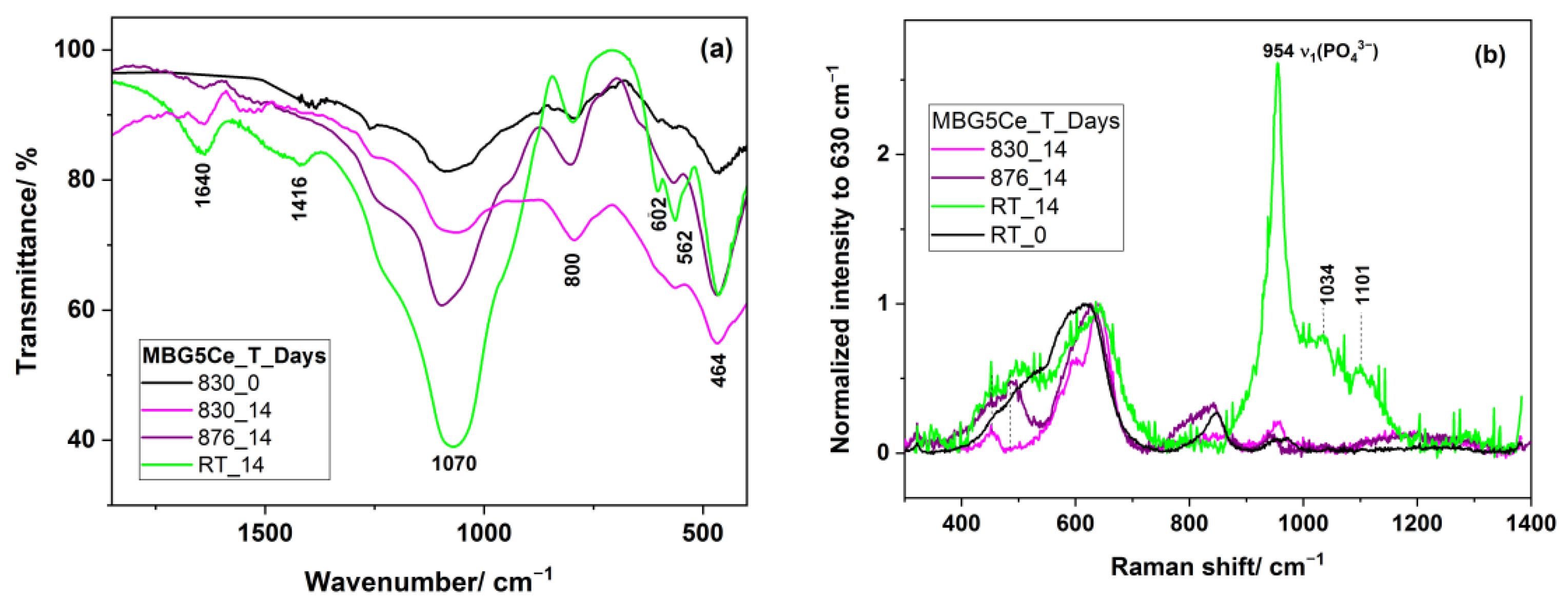
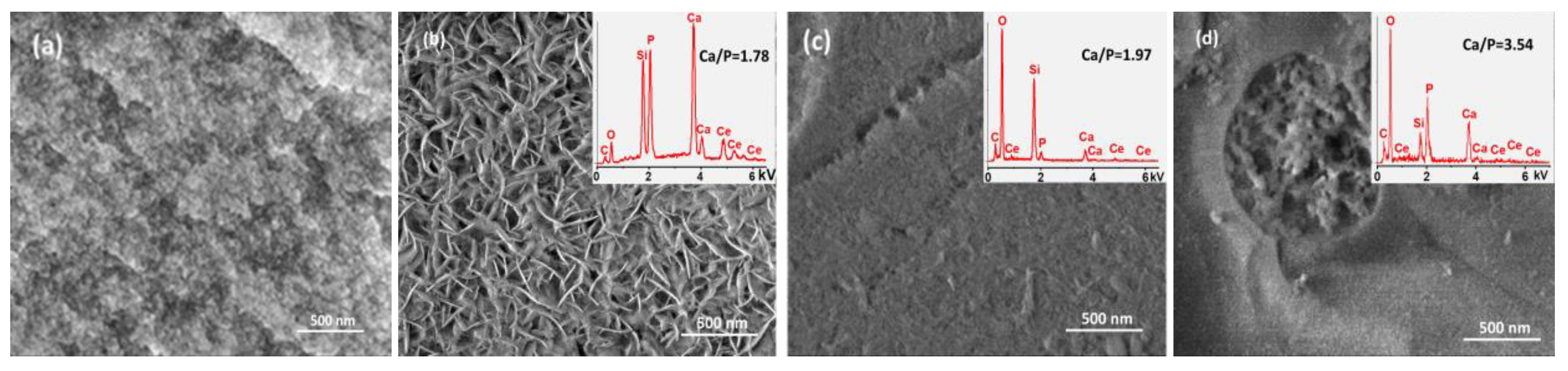
2.5. Bioactivity of the MBG5Ce_T
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Sol–Gel Preparation Methods
4.3. Bioactivity of the Devitrified Glasses
4.4. Characterization
4.4.1. Thermal Characterization
4.4.2. Structural Characterization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clupper, D.C.; Hench, L.L. Crystallization kinetics of tape cast bioactive glass 45S5. J. Non-Cryst. Solids 2003, 318, 43–48. [Google Scholar] [CrossRef]
- Jones, J.R. Reprint of: Review of bioactive glass: From Hench to hybrids. Acta Biomater. 2013, 9, 4457–4486. [Google Scholar] [CrossRef] [PubMed]
- Filho, O.P.; LaTorre, G.P.; Hench, G.P. Effect of crystallization on apatite-layer formation of bioactive glass 45S5. J. Biomed. Mater. Res. 1996, 30, 509–514. [Google Scholar] [CrossRef]
- Crovacea, M.C.; Soares, V.O.; Rodrigues, A.C.M.; Peitl, O.; Raucci, L.M.S.C.; Oliveira, P.T.; Zanotto, E.D. Understanding the mixed alkali effect on the sinterability and in vitro performance of bioactive glasses. J. Eur. Ceram. Soc. 2021, 41, 4391–4405. [Google Scholar] [CrossRef]
- Atkinson, I.; Anghel, E.M.; Predoana, L.; Mocioiu, O.C.; Jecu, L.; Raut, I.; Munteanu, C.; Culita, D.; Zaharescu, M. Influence of ZnO addition on the structural, in vitro behavior and antimicrobial activity of sol–gel derived CaO–P2O5–SiO2 bioactive glasses. Ceram. Int. 2016, 42, 3033–3045. [Google Scholar] [CrossRef]
- Deliormanli, A.M. Synthesis and characterization of cerium- and gallium-containing borate bioactive glass scaffolds for bone tissue engineering. J. Mater. Sci. Mater. Med. 2015, 26, 67. [Google Scholar] [CrossRef]
- Pazarçeviren, A.E.; Tahmasebifar, A.; Tezcaner, A.; Keskin, D.; Evis, Z. Investigation of bismuth doped bioglass/graphene oxide nanocomposites for bone tissue engineering. Ceram. Int. 2018, 44, 3791–3799. [Google Scholar] [CrossRef]
- Siqueira, L.; Campos, T.M.B.; Camargo, S.E.A.; Thim, G.P.; Triches, E.S. Structural, crystallization and cytocompatibility evaluation of the 45S5 bioglass-derived glass-ceramic containing niobium. J. Non-Cryst. Solids 2021, 555, 120629. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhang, Y.; Montazerian, M.; Gulbiten, O.; Mauro, J.C.; Zanotto, E.D.; Yue, Y. Understanding Glass through Differential Scanning Calorimetry. Chem. Rev. 2019, 119, 7848–7939. [Google Scholar] [CrossRef]
- Bellucci, D.; Cannillo, V.; Sola, A. An Overview of the Effects of Thermal Processing on Bioactive Glasses. Sci. Sinter. 2010, 42, 307–320. [Google Scholar] [CrossRef]
- Fagerlund, S.; Masserax, J.; Hupa, L.; Hupa, M. T–T–T behaviour of bioactive glasses 1–98 and 13–93. J. Eur. Ceram. Soc. 2012, 32, 2731–2738. [Google Scholar] [CrossRef]
- Arstila, H.; Vedel, E.; Hupa, L.; Hupa, M. Factors affecting crystallization of bioactive glasses. J. Eur. Ceram. Soc. 2007, 27, 1543–1546. [Google Scholar] [CrossRef]
- Nawaz, Q.; Pablos-Martín, A.; Silva, J.M.S.; Hurle, K.; Contreras-Jaimes, A.T.; Brauer, D.S.; Boccaccini, A.R. New insights into the crystallization process of sol-gel–derived 45S5 bioactive glass. J. Am. Ceram. Soc. 2020, 103, 4234–4247. [Google Scholar] [CrossRef]
- Wetzel, R.; Blochberger, M.; Scheffler, F.; Hupa, L.; Brauer, D.S. Mg or Zn for Ca substitution improves the sintering of bioglass 45S5. Sci. Rep. 2020, 10, 15964. [Google Scholar] [CrossRef]
- Schumacher, M.; Habibovic, P.; Rijt, S. Mesoporous bioactive glass composition effects on degradation and bioactivity. Bioact. Mater. 2021, 6, 1921–1931. [Google Scholar] [CrossRef]
- Brinker, C.J.; Lu, Y.; Sellinger, A.; Fan, H. Evaporation-induced self-assembly: Nanostructures madeeasy. Adv. Mater. 1999, 11, 579–585. [Google Scholar] [CrossRef]
- Lalzawmliana, V.; Anand, B.; Roy, M.; Kundu, B.; Nandi, S.K. Mesoporous bioactive glasses for bone healing and biomolecules delivery. Mater. Sci. Eng. C 2022, 106, 110180. [Google Scholar] [CrossRef]
- Baino, F.; Fiume, E. 3D Printing of Hierarchical Scaffolds Based on Mesoporous Bioactive Glasses (MBGs)—Fundamentals and Applications. Materials 2020, 13, 1688. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Yu, C.; Zhou, X.; Tang, J.; Zhao, D. Highly Ordered Mesoporous Bioactive Glasses with Superior In Vitro Bone-Forming Bioactivities. Angew. Chem. Int. Ed. 2004, 43, 5980–5984. [Google Scholar] [CrossRef]
- Wen, C.; Bai, N.; Luo, L.; Ye, J.; Zhan, X.; Zhang, Y.; Sa, B. Structural behaviorand in vitro bioactivity evaluation of hydroxyapatite-like bioactive glass based on the SiO2-CaO-P2O5system. Ceram. Int. 2021, 47, 18094–18104. [Google Scholar] [CrossRef]
- Baranowska, A.; Lesniak, M.; Kochanowicz, M.; Zmojda, J.; Miluski, P.; Dorosz, D. Crystallization Kinetics and Structural Properties of the 45S5 Bioactive Glass and Glass-Ceramic Fiber Doped with Eu3+. Materials 2020, 13, 1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kissinger, H.E. Reaction kinetics in differential thermal analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Vyazovkin, S. Kissinger Method in Kinetics of Materials: Things to Beware and Be Aware of. Molecules 2020, 25, 2813. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, H.; Serra, J.; González, P.; León, B. Structural study of sol–gel silicate glasses by IR and Raman spectroscopies. J. Non-Cryst. Solid 2009, 355, 475–480. [Google Scholar] [CrossRef]
- Macon, A.L.B.; Lee, S.; Poologasundarampillai, G.; Kasuga, T.; Jones, J.R. Synthesis and dissolution behaviour of CaO/SrO-containing sol–gel-derived 58S glasses. J. Mater. Sci. 2017, 52, 8858–8870. [Google Scholar] [CrossRef]
- Martin, R.A.; Yue, S.; Hanna, J.V.; Lee, P.D.; Newport, R.J.; Smith, M.E.; Jones, J.R. Characterizing the hierarchical structures of bioactive sol–gel silicate glass and hybrid scaffolds for bone regeneration. Philos. Trans. R. Soc. A 2012, 370, 1422–1443. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Dell’Era, A.; Ciprioti, S.V. Synthesis, structural, spectroscopic and thermoanalytical study of sol–gel derived SiO2–CaO–P2O5 gel and ceramic materials. Thermochim. Acta 2016, 625, 20–27. [Google Scholar] [CrossRef]
- Innocenzi, P. Infrared spectroscopy of sol–gel derived silica-based films: A spectra-microstructure overview. J. Non-Cryst. Solids 2003, 316, 309–319. [Google Scholar] [CrossRef]
- Ianoul, A.; Coleman, T.; Asher, S.A. UV Resonance Raman Spectroscopic Detection ofNitrate and Nitrite in Wastewater Treatment Processes. Anal. Chem. 2002, 74, 1458–1461. [Google Scholar] [CrossRef]
- Brooker, M.H. Raman and i.r. spectra of zinc, cadmium and calcium nitrate: A study of the low temperature phase transitions in calcium nitrate. Spectrochim. Acta 1975, 32, 369–377. [Google Scholar] [CrossRef]
- Zhu, F.; Zhou, H.; Zhou, Y.; Ge, H.; Liu, H.; Fang, C.; Fang, Y. Ab Initio Investigation of the Micro-species and Raman Spectra in Ca(NO3)2Solution. J. Clust. Sci. 2017, 28, 2293–2307. [Google Scholar] [CrossRef]
- Todan, T.; Anghel, E.M.; Osiceanu, P.; Turcu, R.V.F.; Atkinson, I.; Simon, S.; Zaharescu, M. Structural characterization of some sol–gel derived phosphosilicate glasses. J. Mol. Struct. 2015, 1086, 161–171. [Google Scholar] [CrossRef]
- Atkinson, I.; Anghel, E.M.; Petrescu, S.; Seciu, A.M.; Stefan, L.M.; Mocioiu, O.C.; Predoana, L.; Voicescu, M.; Somacescu, S.; Culita, D.; et al. Cerium-containing mesoporous bioactive glasses: Material characterization, in vitro bioactivity, biocompatibility and cytotoxicity evaluation. Micropor. Mesopor. Mat. 2019, 276, 76–88. [Google Scholar] [CrossRef]
- Buriti, J.S.; Barreto, M.E.V.; Barbosa, F.C.; Buriti, B.M.A.B.; Souza, J.W.L.; Pina, H.V.; Rodrigues, P.L.; Fook, M.V.L. Synthesis and characterization of Ag-doped 45S5 bioglass and chitosan/45S5-Ag biocomposites for biomedical applications. J. Therm. Anal. Calorim. 2021, 145, 39–50. [Google Scholar] [CrossRef]
- Martınez, A.; Izquierdo-Barba, I.; Vallet-Regı, M. Bioactivity of a CaO-SiO2 Binary Glasses System. Chem. Mater. 2000, 12, 3080–3088. [Google Scholar] [CrossRef]
- Siqueira, R.L.; Zanotto, E.D. The influence of phosphorus precursors on the synthesis and bioactivity of SiO2–CaO–P2O5 sol–gel glasses and glass–ceramics. J. Mater. Sci: Mater. Med. 2013, 24, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Cai, S.; Xu, G.; Ren, M.; Wang, X.; Zhang, R.; Niu, S.; Zhao, H. Sol–gel derived mesoporous 58S bioactive glass coatings on AZ31 magnesium alloy and in vitro degradation behavior. Surf. Coat. Technol. 2014, 240, 137–144. [Google Scholar] [CrossRef]
- Zaharescu, M.; Predoana, L.; Pandele, J. Relevance of thermal analysis for sol–gel-derived nanomaterials. J. Sol-Gel Sci. Technol. 2018, 86, 7–23. [Google Scholar] [CrossRef]
- Jones, J.R.; Ehrenfried, L.M.; Hench, L.L. Optimising bioactive glass scaffolds for bone tissue engineering. Biomaterials 2006, 27, 964–973. [Google Scholar] [CrossRef]
- Likitvanichkul, S.; Lacourse, W.C. Apatite–wollastonite glass-ceramics. Part I Crystallization kinetics by differential thermal analysis. J. Mater. Sci. 1998, 33, 509–5904. [Google Scholar] [CrossRef]
- Notingher, I.; Jones, J.R.; Verrier, S.; Bisson, I.; Embanga, P.; Edwards, P.; Polak, J.M.; Hench, L.L. Application of FTIR and Raman spectroscopy to characterisation of bioactive materials and living cells. Spectroscopy 2003, 17, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Senthilkumar, R.P.; Bhuvaneshwari, V.; Ranjithkumar, R.; Sathiyavimal, S.; Malayaman, V.; Chandarshekar, B. Synthesis, characterization and antibacterial activity of hybrid chitosan-cerium oxide nanoparticles: As a bionanomaterials. Int. J. Biol. Macromol. 2017, 104, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Padilla, S.; Roman, J.; Carenas, A.; Vallet-Regi, M. The influence of the phosphorus content on the bioactivity of sol–gel glass ceramics. Biomaterials 2005, 26, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.M.; Clark, A.E.; Hench, L.L. Calcium phosphate formation on sol-gel-derived bioFactive glasses in vitro. J. Biomed. Mater. Res. 1994, 28, 693–698. [Google Scholar] [CrossRef]
- De Ligny, D.; Neuville, D.R. 12-Raman spectroscopy: A valuable tool to improve our understanding of nucleation and growth mechanism. In From Glass to Crystal. Nucleation, Growth and Phase Separation: From Research to Applications; Neuville, D.R., Cormier, L., Caurant, D., Montagne, L., Eds.; EDP Sciences: Les Ulis, France, 2017; pp. 319–344. [Google Scholar]
- Taniguchi, T.; Watanabe, T.; Sugiyama, N.; Subramani, A.K.; Wagata, H.; Matsushita, N.; Yoshimura, M. Identifying defects in ceria-based nanocrystals by UV resonance Raman spectroscopy. J. Phys. Chem. C 2009, 113, 19789–19793. [Google Scholar] [CrossRef]
- Olivares, M.; Zuluaga, M.C.; Ortega, L.A.; Murelaga, X.; Alonso-Olazabal, A.; Urteaga, M.; Amundaray, L.; Alonso-Martina, I.; Etxebarria, N. Characterisation of fine wall and eggshell Roman pottery by Raman spectroscopy. J. Ram. Spectrosc. 2010, 41, 1543–1549. [Google Scholar] [CrossRef]
- Huang, E.; Chen, C.H.; T. Huang, T.; Lin, E.H.; Xu, J.-A. Raman spectroscopic characteristics of Mg-Fe-Ca pyroxenes. Am. Mineral. 2000, 85, 473–479. [Google Scholar] [CrossRef]
- Uy, D.; O’Neill, A.E.; Xu, L.; Weber, W.H.; McCabe, R.W. Observation of cerium phosphate in aged automotive catalysts using Raman spectroscopy. Appl. Catal. B 2003, 41, 269–278. [Google Scholar] [CrossRef]
- Awonusi, A.; Michael, A.E.; Morris, D.; Tecklenbur, M.M.J. Carbonate Assignment and Calibration in the Raman Spectrum of Apatite. Calcif. Tissue Int. 2007, 81, 46–52. [Google Scholar] [CrossRef]
- Zambon, A.; Malavasi, G.; Pallini, A.; Fraulini, F.; Lusvardi, G. Cerium Containing Bioactive Glasses: A Review. ACS Biomater. Sci. Eng. 2021, 7, 4388–4401. [Google Scholar] [CrossRef]
- Panah, N.G.; Atkin, R.; Sercombe, T.B. Effect of low temperature crystallization on 58S bioactive glass sintering and compressive strength. Ceram. Int. 2021, 47, 30349–30357. [Google Scholar] [CrossRef]
- Martin, M.T.; Maliqi, L.; Keevend, K.; Guimond, S.; Ng, J.; Armagan, E.; Rottmar, M.; Herrmann, I.K. One-Step Synthesis of Versatile Antimicrobial Nano-Architected Implant Coatings for Hard and Soft Tissue Healing. ACS Appl. Mater. Interfaces 2021, 13, 33300–33310. [Google Scholar] [CrossRef]
- Augis, J.A.; Bennett, J.E. Calculation of the Avrami Parameters for Heterogeneous Solid State Reactions Using a Modification of the Kissinger Method. J. Thermal. Anal. Calorim. 1978, 13, 283–292. [Google Scholar] [CrossRef]
- Amorósa, J.E.; Blascoa, E.; Morenoa, A.; Marínc, N.; Feliu, C. Sinter-crystallisation kinetics of a SiO2–Al2O3–CaO–MgO–SrO glass-ceramic glaze. J. Non-Cryst. Solids 2020, 532, 119900. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gel Code | Stage No | ΔT/°C | Mass/% | Assignment | Total Loss/% |
|---|---|---|---|---|---|
| 0Ce | 1 | 25–132 | 6.30 | Physically absorbed water [34,35] | 59.91 |
| 2 | 132–234 | 16.16 | Chemisorbed water and organic oxidation [13] | ||
| 3 | 234–301 | 18.05 | |||
| 4 | 301–497 | 18.93 | Pluronic and nitrate decomposition [26,35] | ||
| 1Ce | 1 | 25–134 | 7.14 | Physically absorbed water | 58.39 |
| 2 | 134–236 | 16.63 | Chemisorbed water and organic oxidation | ||
| 3 | 236–298 | 12.94 | |||
| 4 | 298–390 | 13.85 | Pluronic and nitrate decomposition | ||
| 5 | 390–640 | 6.96 | |||
| 5Ce | 1 | 25–106 | 3.39 | Physically absorbed water | 60.85 |
| 2 | 106–227 | 20.17 | Chemisorbed water and organic oxidation | ||
| 3 | 227–293 | 16.11 | |||
| 4 | 293–620 | 20.04 | Pluronic and nitrate decomposition |
| MBG Code | Tc1 (°C/Phases) | Tc2 (°C/Phases) | Tc3 (°C/Phases) |
|---|---|---|---|
| 0Ce | 878/ | 951/ | 1012/ |
| A (68.01%), W (20.72%) PW (11.27%) | A (68.34%), W (10.04%), PW (21.62%) | A (70.98), W (17.69%), PW (11.33%) | |
| 1Ce | 865/ | 900/ | - |
| A (90.10%), W (9.90%) | A (85.88%), W (15.12%) | ||
| 5Ce | 830/ | 876/ | 906/ |
| A (38.21%), C (14.10%), PW (47.68) | A (54.15%, C (13.41%) | A (59.05%), C (7.80%), PW (33.14%) | |
| PW (32.44%) |
| Sample | Intercept | Slope | Ea | n | R2 |
|---|---|---|---|---|---|
| MBG0Ce | 64.5969 | 87.8985 ± 5.22 | 730.82 ± 43.40 | 1.5979 | 0.9860 |
| MBG1Ce | 64.9096 | 87.2713 ± 3.23 | 725.61 ± 26.85 | 1.8879 | 0.9959 |
| MBG5Ce | −44.7813 | 62.2325 ± 3.17 | 517.43 ± 26.35 | 1.1871 | 0.9922 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anghel, E.M.; Petrescu, S.; Mocioiu, O.C.; Cusu, J.P.; Atkinson, I. Influence of Ceria Addition on Crystallization Behavior and Properties of Mesoporous Bioactive Glasses in the SiO2–CaO–P2O5–CeO2 System. Gels 2022, 8, 344. https://doi.org/10.3390/gels8060344
Anghel EM, Petrescu S, Mocioiu OC, Cusu JP, Atkinson I. Influence of Ceria Addition on Crystallization Behavior and Properties of Mesoporous Bioactive Glasses in the SiO2–CaO–P2O5–CeO2 System. Gels. 2022; 8(6):344. https://doi.org/10.3390/gels8060344
Chicago/Turabian StyleAnghel, Elena Maria, Simona Petrescu, Oana Catalina Mocioiu, Jeanina Pandele Cusu, and Irina Atkinson. 2022. "Influence of Ceria Addition on Crystallization Behavior and Properties of Mesoporous Bioactive Glasses in the SiO2–CaO–P2O5–CeO2 System" Gels 8, no. 6: 344. https://doi.org/10.3390/gels8060344
APA StyleAnghel, E. M., Petrescu, S., Mocioiu, O. C., Cusu, J. P., & Atkinson, I. (2022). Influence of Ceria Addition on Crystallization Behavior and Properties of Mesoporous Bioactive Glasses in the SiO2–CaO–P2O5–CeO2 System. Gels, 8(6), 344. https://doi.org/10.3390/gels8060344