

Highly Thermally Resistant Bisamide Gelators as Pharmaceutical Crystallization Media

 ,
,  ,
, 

Abstract
:
1. Introduction
2. Results and Discussion
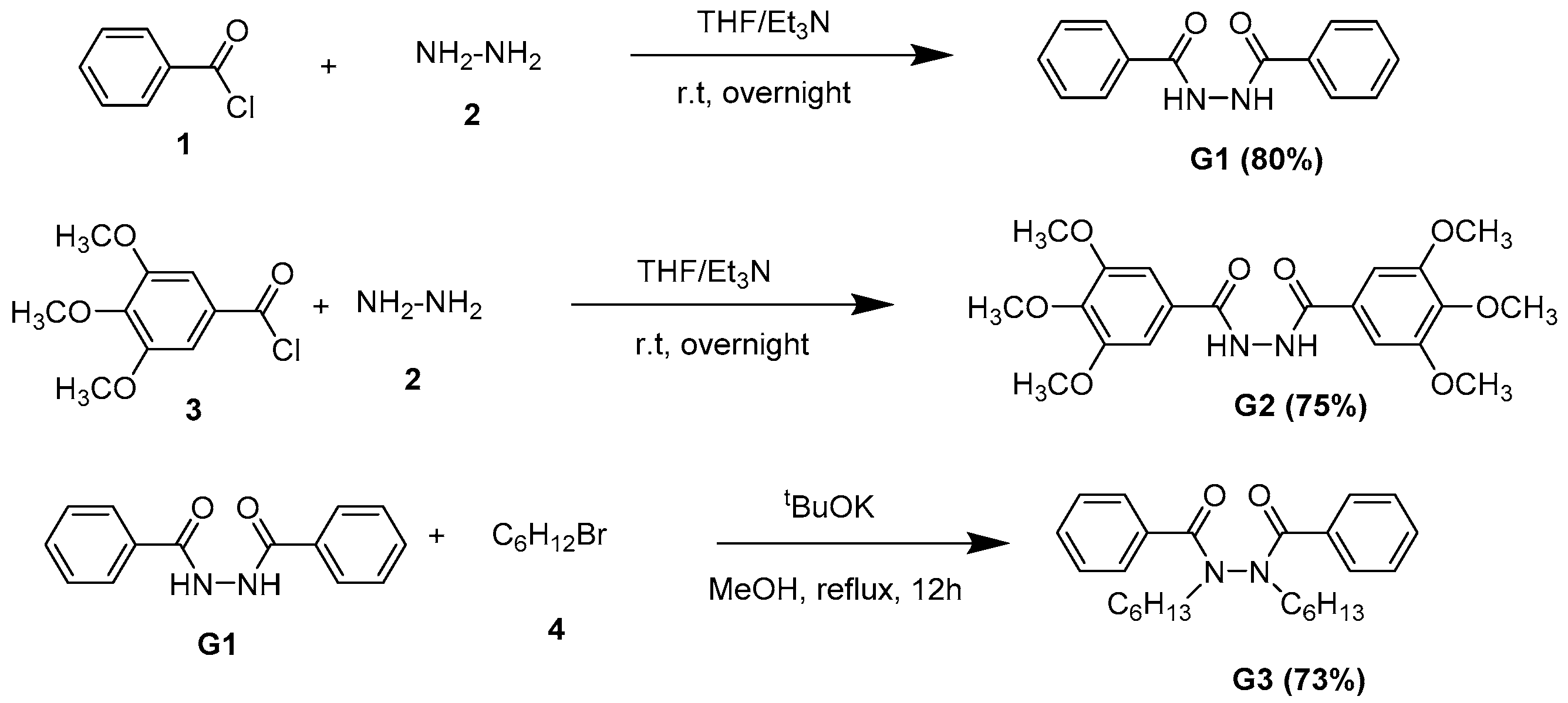
2.1. Synthetic Procedure
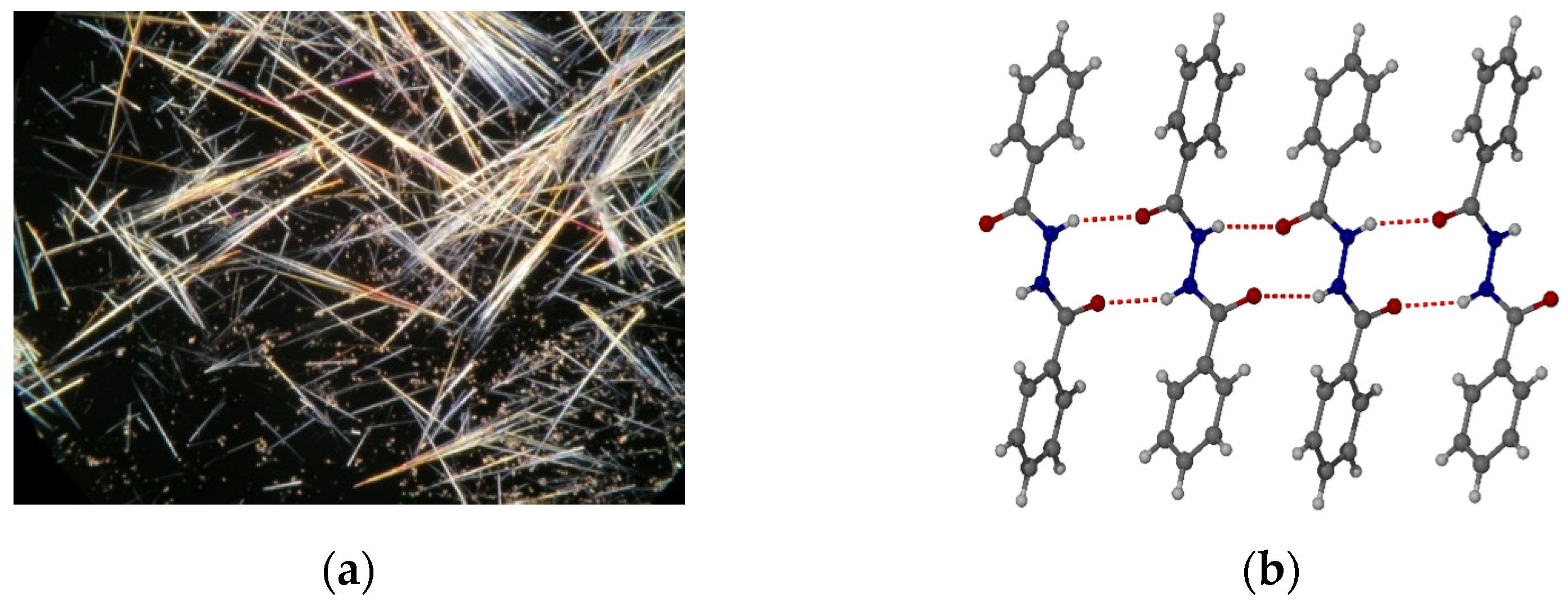
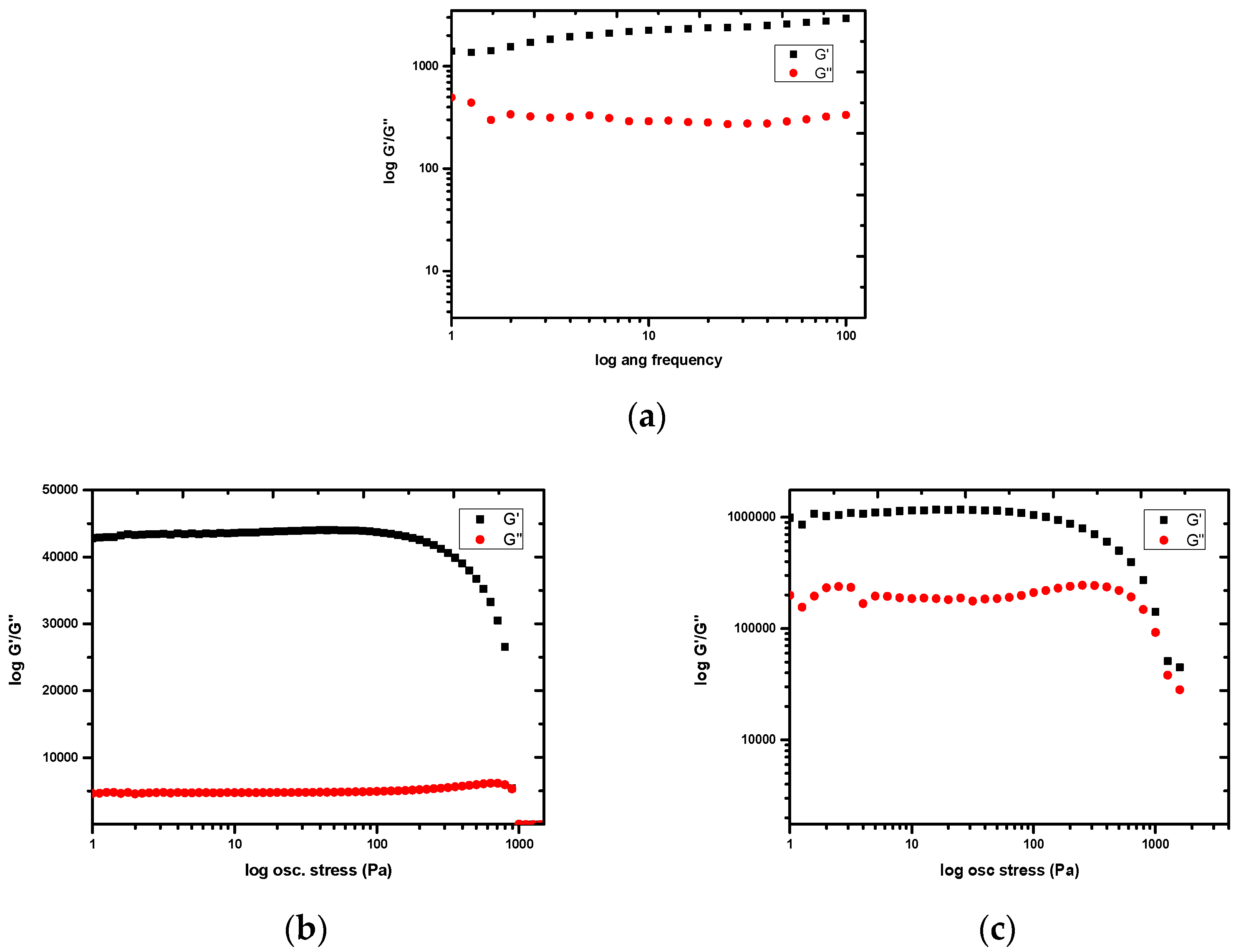
2.2. Gelation Tests
2.3. Stimuli Response
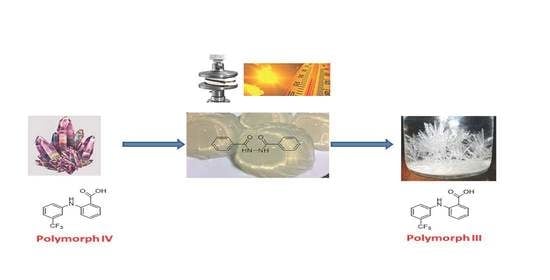
2.4. Gel Phase Crystallization of Pharmaceutical Drugs
3. Conclusions
4. Materials and Methods
Synthetic Procedures for the Synthesis of G1–3
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jordan Lloyd, D. Colloid Chemistry. Alexander, J., Ed.; The Chemical Catalogue Company: New York, NY, USA, 1926; Volume 1, pp. 767–782. [Google Scholar]
- Terech, P.; Weiss, R.G. Low Molecular Mass Gelators of Organic Liquids and the Properties of Their Gels. Chem. Rev. 1997, 97, 3133–3159. [Google Scholar] [CrossRef] [PubMed]
- Draper, E.R.; Adams, D.J. Low-Molecular-Weight Gels: The State of the Art. Chem 2017, 3, 390–410. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Corradini, M.G.; Weiss, R.G.; Raghavan, S.R.; Rogers, M.A. To gel or not to gel: Correlating molecular gelation with solvent parameters. Chem. Soc. Rev. 2015, 44, 6035–6058. [Google Scholar] [CrossRef]
- Dastidar, P. Designing Supramolecular Gelators: Challenges, Frustrations, and Hopes. Gels 2019, 5, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirst, A.R.; Coates, I.A.; Boucheteau, T.R.; Miravet, J.F.; Escuder, B.; Castelletto, V.; Hamley, I.W.; Smith, D.K. Low-molecular-weight gelators: Elucidating the principles of gelation based on gelator solubility and a cooperative self-assembly model. J. Am. Chem. Soc. 2008, 130, 9113–9121. [Google Scholar] [CrossRef]
- Yu, G.; Yan, X.; Han, C.; Huang, F. Characterization of supramolecular gels. Chem. Soc. Rev. 2013, 42, 6697–6722. [Google Scholar] [CrossRef]
- Foster, J.A.; Damodaran, K.K.; Maurin, A.; Day, G.M.; Thompson, H.P.G.; Cameron, G.J.; Bernal, J.C.; Steed, J.W. Pharmaceutical polymorph control in a drug-mimetic supramolecular ge. Chem. Sci. 2017, 8, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Foster, J.A.; Piepenbrock, M.O.M.; Lloyd, G.O.; Clarke, N.; Howard, J.A.K.; Steed, J.W. Anion-switchable supramolecular gels for controlling pharmaceutical crystal growth. Nat. Chem. 2010, 2, 1037–1043. [Google Scholar] [CrossRef]
- Aparicio, F.; Matesanz, E.; Sanchez, L. Cooperative self-assembly of linear organogelators. Amplification of chirality and crystal growth of pharmaceutical ingredients. Chem. Commun. 2012, 48, 5757–5759. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker, R. Polymorphism in the Pharmaceutical Industry; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Lee, E.H. Asian A practical guide to pharmaceutical polymorph screening & selection. J. Pharm. Sci. 2014, 9, 163–175. [Google Scholar] [CrossRef]
- Dawn, A.; Andrew, K.S.; Yufit, D.S.; Hong, Y.X.; Reddy, J.P.; Jones, C.D.; Aguilar, J.A.; Steed, J.W. Supramolecular Gel Control of Cisplatin Crystallization: Identification of a New Solvate Form Using a Cisplatin-Mimetic Gelator. Growth Des. 2015, 15, 4591–4599. [Google Scholar] [CrossRef] [Green Version]
- Buendía, J.; Matesanz, E.; Smith, D.K.; Sánchez, L. Multi-component supramolecular gels for the controlled crystallization of drugs: Synergistic and antagonistic effects. CrystEngComm 2015, 17, 8146–8152. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.K.; Steed, J.W. Supramolecular gel phase crystallization: Orthogonal self-assembly under non-equilibrium conditions. Chem. Soc. Rev. 2014, 43, 2080–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.R.; Sun, S.-S. Supramolecular Assemblies of Amide-Derived Organogels Featuring Rigid π-Conjugated Phenylethynyl Frameworks. Langmuir 2013, 29, 15146–15158. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Sundarajan, P.R. Low Molecular Weight Organogels Based on Long-Chain Carbamates. Langmuir 2005, 21, 3802–3807. [Google Scholar] [CrossRef] [PubMed]
- Shanmuga Sundara Raj, S.; Yamin, B.M.; Boshaala, A.M.A.; Tarafder, M.T.H.; Crouse, K.A.; Fun, H.-K. 1,2-Dibenzoylhydrazine. Acta Cryst. 2000, 56, 1011–1012. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.M.; Venkataramana, P.; Swamy, P.Y.; Chityala, Y. N-Amino-1,8-Naphthalimide is a Regenerated Protecting Group for Selective Synthesis of Mono-N-Substituted Hydrazines and Hydrazides. Chem. Eur. J. 2021, 27, 17713–17721. [Google Scholar] [CrossRef]
- Ekiz, F.; Rende, E.; Timur, S.; Toppare, L. A novel functional conducting polymer: Synthesis and application to biomolecule immobilization. J. Mater. Chem. 2012, 22, 22517–22525. [Google Scholar] [CrossRef]
- Pekcan, O.; Kara, S. Gelation Mechanisms. Mod. Phys. Lett. B 2012, 26, 30019–30046. [Google Scholar] [CrossRef]
- Mukkamala, R.; Weiss, R.G. Physical Gelation of Organic Fluids by Anthraquinone−Steroid-Based Molecules. Structural Features Influencing the Properties of Gels. Langmuir 1996, 12, 1474–1482. [Google Scholar] [CrossRef]
- Takahashi, A.; Sakai, M.; Kato, T. Melting Temperature of Thermally Reversible Gel. VI. Effect of Branching on the Sol–Gel Transition of Polyethylene Gels. Polym. J. 1980, 12, 335–341. [Google Scholar] [CrossRef]
- Byrne, P.; Lloyd, G.O.; Applegarth, L.; Anderson, K.M.; Clarke, N.; Steed, J.W. Metal-induced gelation in dipyridylureas. New J. Chem. 2010, 34, 2261–2274. [Google Scholar] [CrossRef]
- Song, S.; Wang, L.; Yao, C.; Wang, Z.; Xie, G.; Tao, X. Crystallization of Sulfathiazole in Gel: Polymorph Selectivity and Cross-Nucleation. Cryst. Growth Des. 2020, 20, 9–16. [Google Scholar] [CrossRef]
- Ran, X.; Shi, L.; Zhang, K.; Lou, J.; Liu, B.; Guo, L. The Gelation Ability and Morphology Study of Organogel System Based on Calamitic Hydrazide Derivatives. J. Nanomater. 2015, 2015, 357875. [Google Scholar] [CrossRef] [Green Version]
- Malkin, A.Y.; Isayev, A.I. Rheology: Concepts, Methods and Applications; Chemtech Publishing: Toronto, ON, Canada, 2006. [Google Scholar]
- Jin, C.; Song, W.J.; Liu, T.; Xin, J.N.; Hiscox, W.C.; Zhang, J.W.; Liu, G.F.; Kong, Z.W. Temperature and pH Responsive Hydrogels Using Methacrylated Lignosulfonate Cross-Linker: Synthesis, Characterization, and Properties. ACS Sustain. Chem. Eng. 2018, 6, 1763–1771. [Google Scholar] [CrossRef]
- Khaltab, T.A.; Tiu, B.D.B.; Adas, S.; Bunge, S.D.; Advincula, R.C. pH triggered smart organogel from DCDHF-Hydrazone molecular switc. Dyes Pigment. 2016, 130, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Matsuoka, M. Solid-State Polymorphic Transition of Theophylline Anhydrate and Humidity Effect. Cryst. Growth Des. 2007, 7, 411–415. [Google Scholar] [CrossRef]
- Seton, L.; Khamar, D.; Bradshaw, I.J.; Hutcheon, G.A. Solvent-Mediated Central Metals Transformation from a Tetranuclear NiII Cage to a Decanuclear CuII “Pocket”. Cryst. Growth Des. 2010, 10, 3879–3886. [Google Scholar] [CrossRef]
- Patel, M.K.; Tailor, S.M.; Patel, V.H. DFT study and Hirshfeld surface analysis of third polymorph of sulfamerazine. AIP Conf. Proc. 2016, 1728, 020257. [Google Scholar] [CrossRef]
- Munroe, A.; Rasmuson, A.C.; Modnett, B.K.; Croker, D.M. Relative Stabilities of the Five Polymorphs of Sulfathiazole. Cryst. Growth Des. 2012, 12, 2825–2835. [Google Scholar] [CrossRef]
- Anwar, J.; Tarling, S.E.; Barnes, P. Polymorphism of Sulfathiazole. J. Pharm. Sci. 1989, 78, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Fellah, N.; López-Mejías, V.; Ward, M.D. Polymorphic Phase Transformation Pathways under Nanoconfinement: Flufenamic Acid. Cryst. Growth Des. 2020, 20, 7098–7103. [Google Scholar] [CrossRef]
- Saha, S.; Desiraju, G.R. Acid···Amide Supramolecular Synthon in Cocrystals: From Spectroscopic Detection to Property Engineering. J. Am. Chem. Soc. 2018, 140, 6361–6373. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Terekhova, I.V.; Bauer-Brandl, A.; Perlovich, G.L. Thermodynamic and Structural Aspects of Some Fenamate Molecular Crystals. Cryst. Growth Des. 2009, 9, 3265–3272. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta. Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- López-Mejías, V.; Kampf, J.W.; Matzger, A.J. Nonamorphism in flufenamic acid and a new record for a polymorphic compound with solved structures. J. Am. Chem. Soc. 2012, 134, 9872–9875. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | CGC (%wt.) |
|---|---|
| Dichloromethane | 0.6 |
| Ethanol | 1.5 |
| 1-Pentanol | 0.8 |
| 1-Propanol | 0.8 |
| 2-Butanol | 1.3 |
| Acetone | 1.2 |
| Acetonitrile | 1.5 |
| Water | 1.0 |
| Ethyl acetate | 0.6 |
| Solvent | Tsol (°C) (2% wt.) | Tsol (°C) (1% wt.) |
|---|---|---|
| Dichloromethane | 66 | 70 |
| Ethanol | 82 | --- |
| 1-Pentanol | 110 | 78 |
| 1-Propanol | 62 | 63 |
| 2-Butanol | 105 | --- |
| Acetone | 94 | --- |
| Acetonitrile | 93 | --- |
| Water | 121 | 72 |
| Ethyl acetate | 132 | 74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Moya, I.; Sánchez, A.; Saikia, B.; Yufit, D.S.; Prieto, P.; Carrillo, J.R.; Steed, J.W. Highly Thermally Resistant Bisamide Gelators as Pharmaceutical Crystallization Media. Gels 2023, 9, 26. https://doi.org/10.3390/gels9010026
Torres-Moya I, Sánchez A, Saikia B, Yufit DS, Prieto P, Carrillo JR, Steed JW. Highly Thermally Resistant Bisamide Gelators as Pharmaceutical Crystallization Media. Gels. 2023; 9(1):26. https://doi.org/10.3390/gels9010026
Chicago/Turabian StyleTorres-Moya, Iván, Abelardo Sánchez, Basanta Saikia, Dmitry S. Yufit, Pilar Prieto, José Ramón Carrillo, and Jonathan W. Steed. 2023. "Highly Thermally Resistant Bisamide Gelators as Pharmaceutical Crystallization Media" Gels 9, no. 1: 26. https://doi.org/10.3390/gels9010026
APA StyleTorres-Moya, I., Sánchez, A., Saikia, B., Yufit, D. S., Prieto, P., Carrillo, J. R., & Steed, J. W. (2023). Highly Thermally Resistant Bisamide Gelators as Pharmaceutical Crystallization Media. Gels, 9(1), 26. https://doi.org/10.3390/gels9010026