
Probing the Molecular Mechanism of Viscoelastic Relaxation in Transient Networks

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Relationships between Diffusion and Viscoelasticity of Associative Polymer Networks
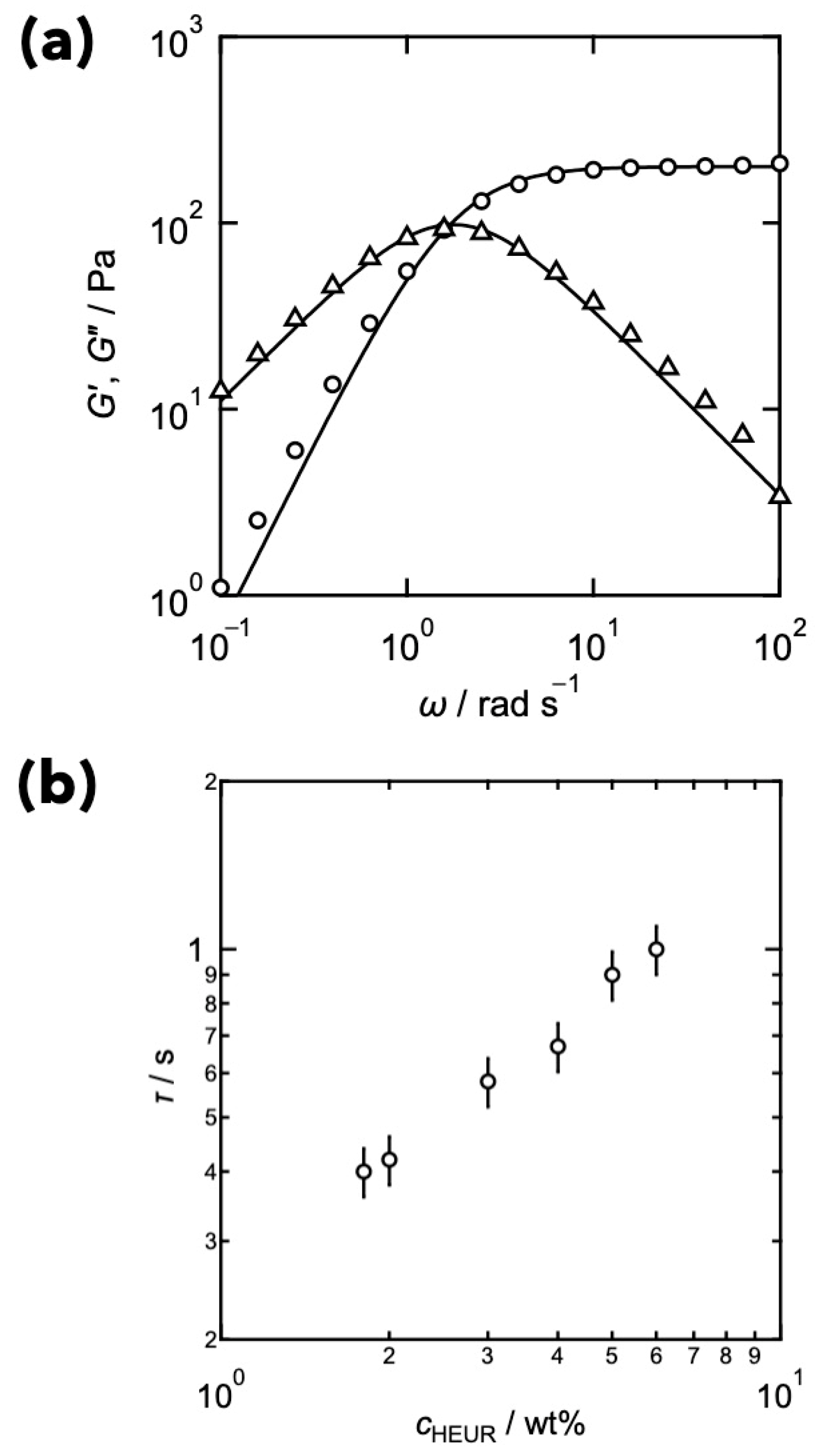
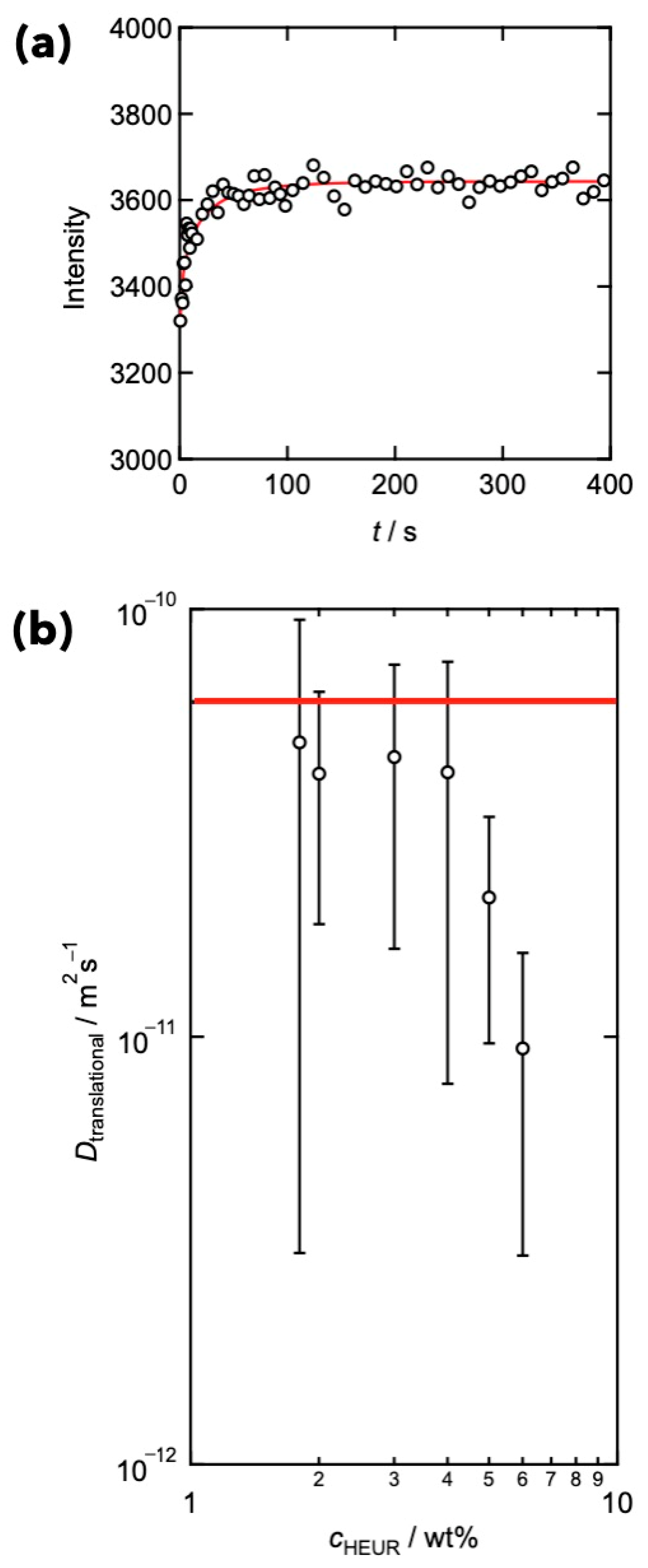
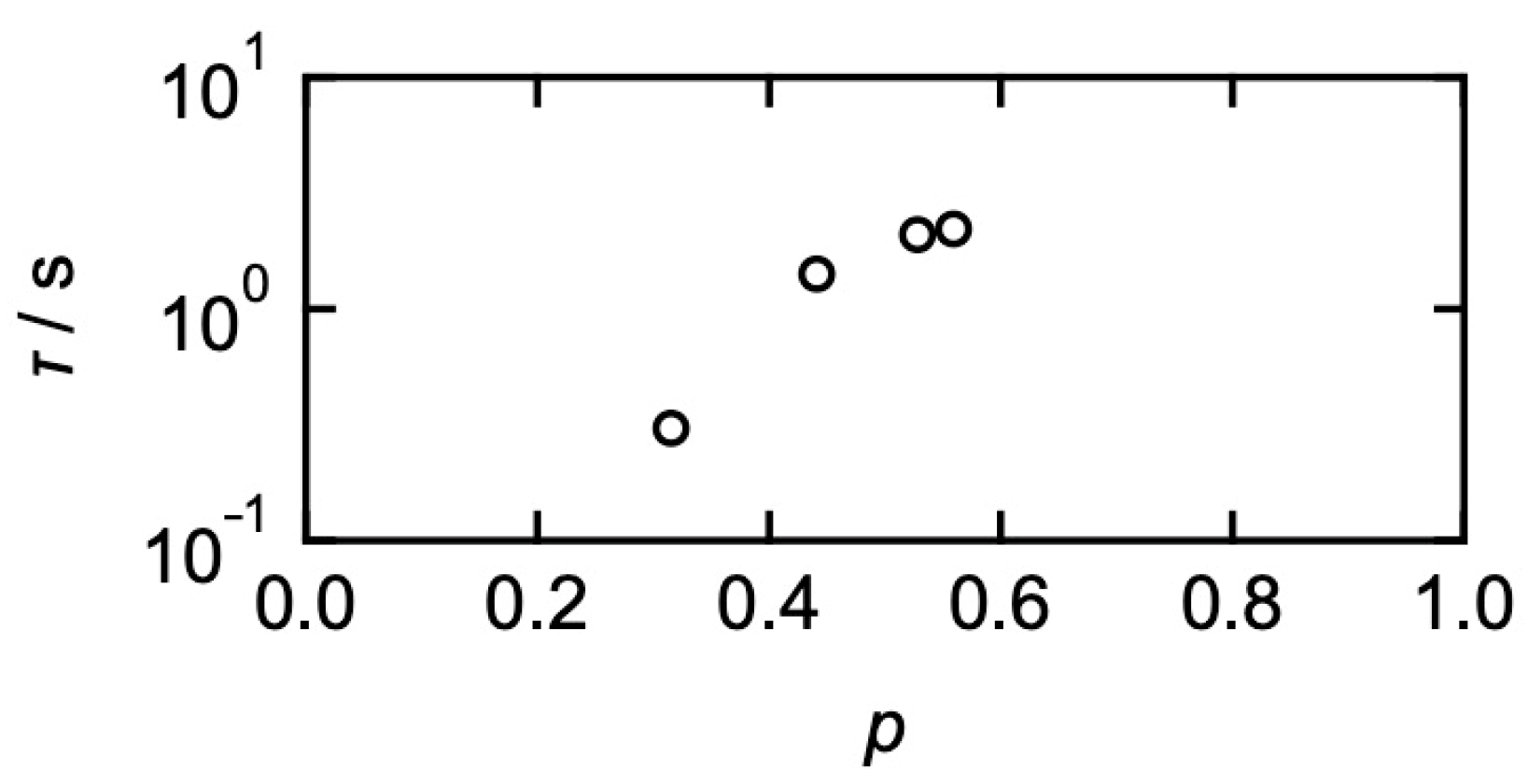
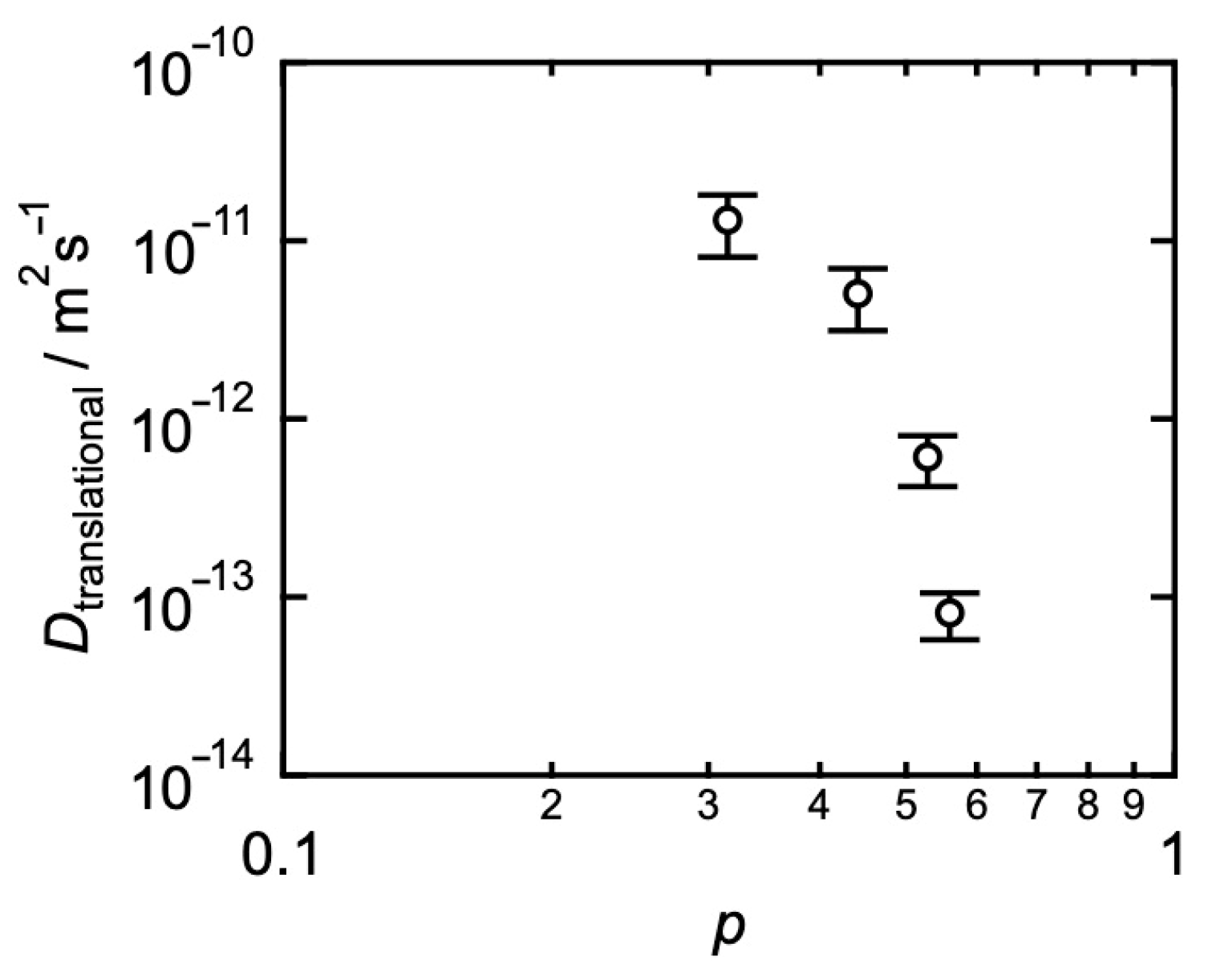
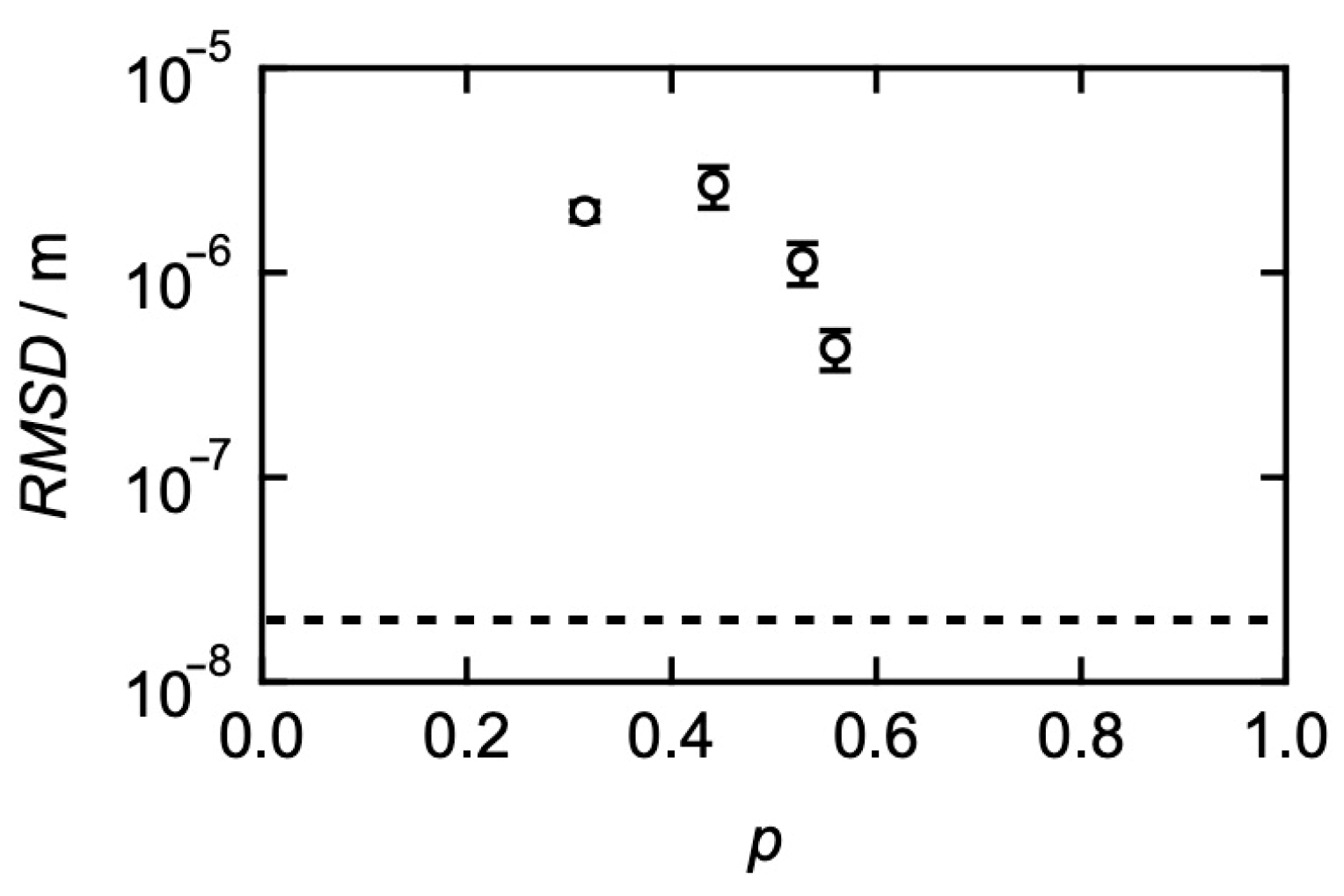
3. Relationships between Translational Diffusion and Viscoelasticity in Transient Networks with Controlled Network Structures
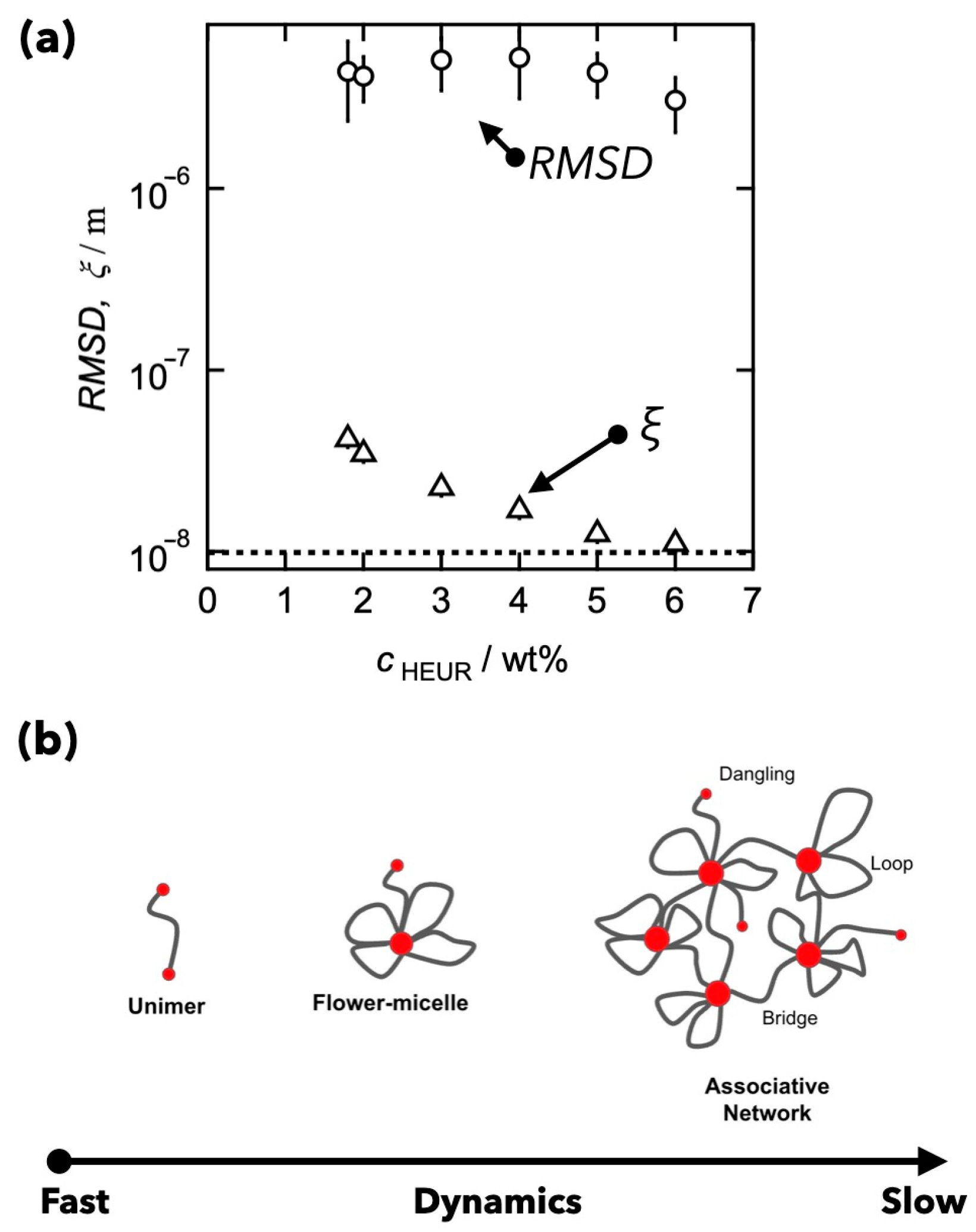
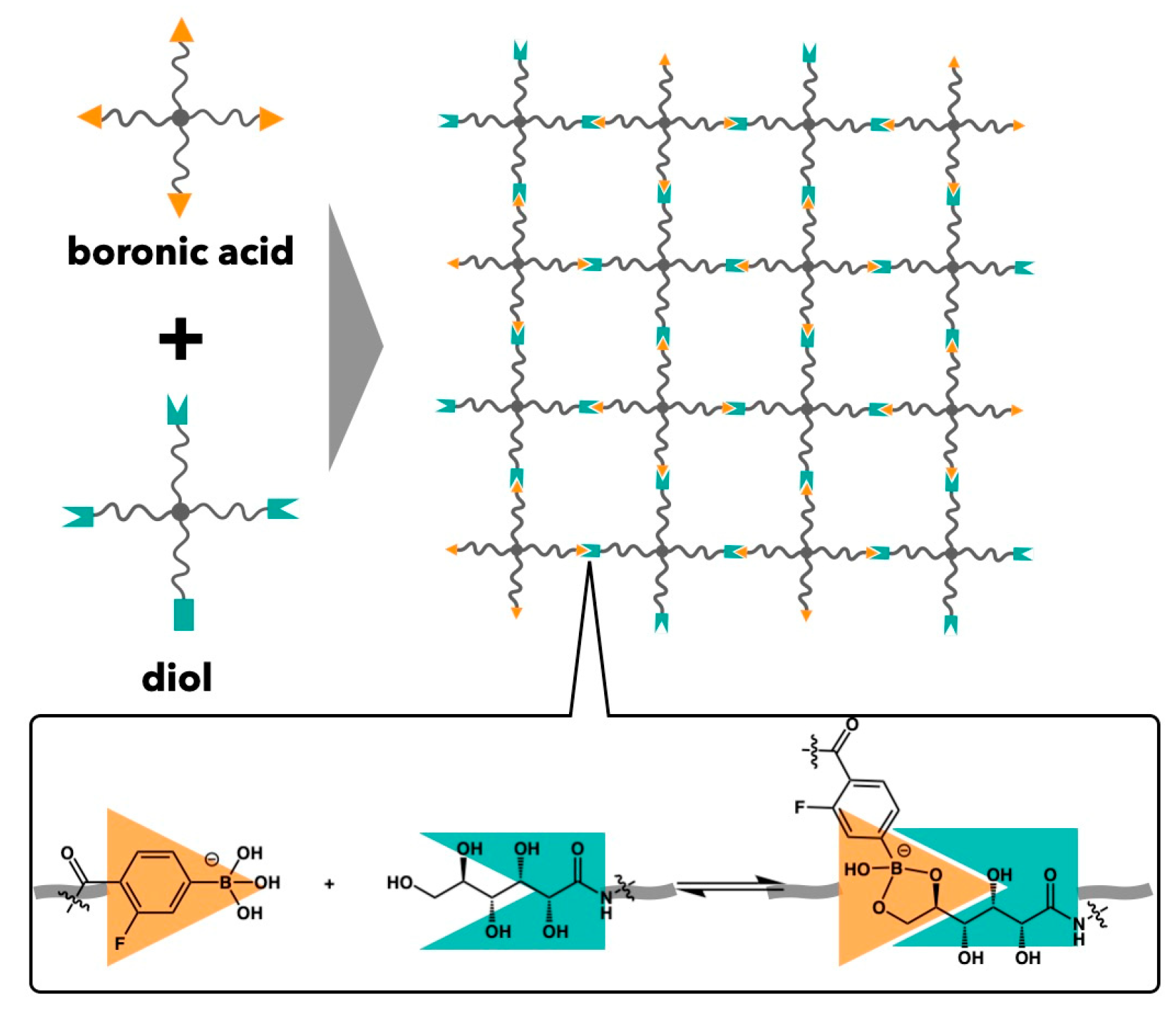
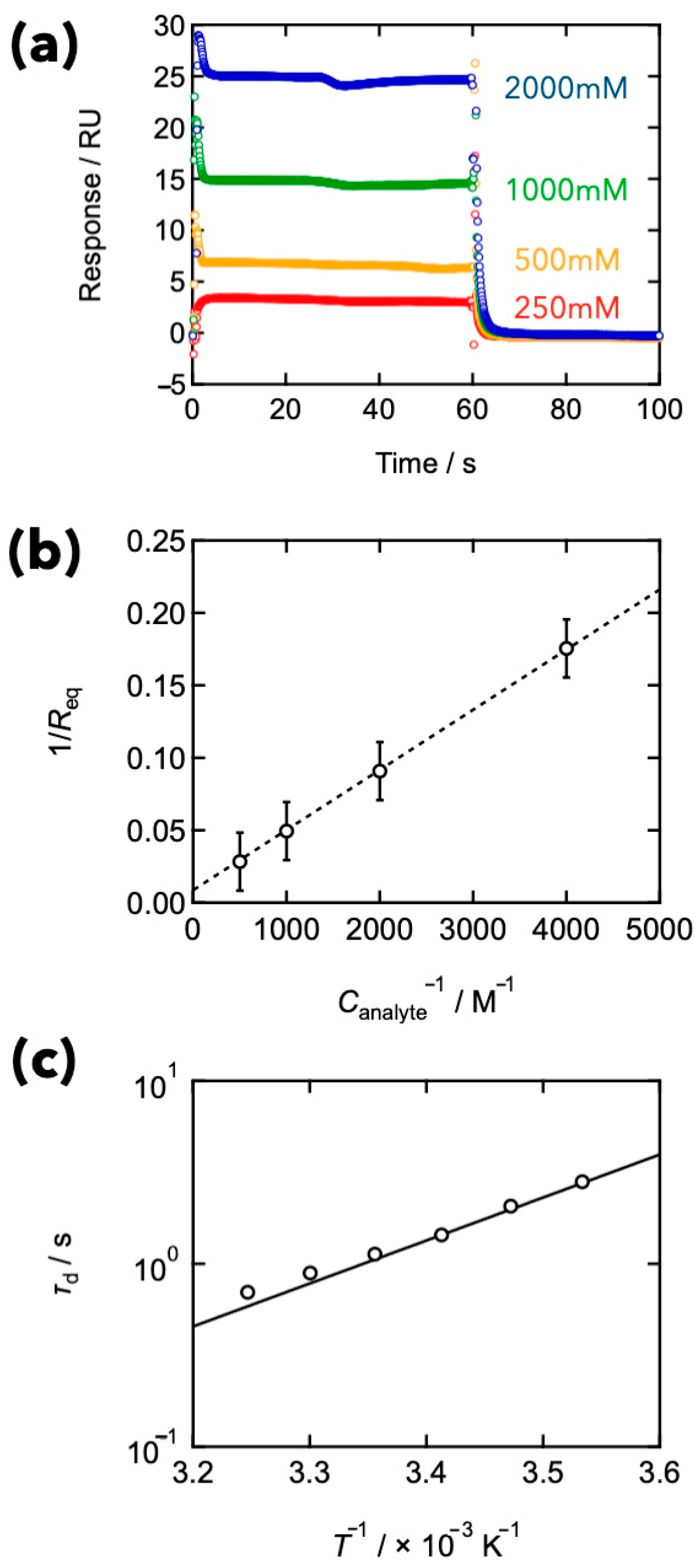
4. Comparison between Binding and Dissociation Kinetics of Crosslinks and Viscoelasticity in Transient Networks with Controlled Network Structures
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-Healing and Thermoreversible Rubber from Supramolecular Assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Bastings, M.M.C.; Koudstaal, S.; Kieltyka, R.E.; Nakano, Y.; Pape, A.C.H.; Feyen, D.A.M.; van Slochteren, F.J.; Doevendans, P.A.; Sluijter, J.P.G.; Meijer, E.W.; et al. A Fast PH-Switchable and Self-Healing Supramolecular Hydrogel Carrier for Guided, Local Catheter Injection in the Infarcted Myocardium. Adv. Healthc. Mater. 2014, 3, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Kouwer, P.H.J.; Koepf, M.; Le Sage, V.A.A.; Jaspers, M.; van Buul, A.M.; Eksteen-Akeroyd, Z.H.; Woltinge, T.; Schwartz, E.; Kitto, H.J.; Hoogenboom, R.; et al. Responsive Biomimetic Networks from Polyisocyanopeptide Hydrogels. Nature 2013, 493, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Rizov, V. Effects of Periodic Loading on Longitudinal Fracture in Viscoelastic Functionally Graded Beam Structures. J. Comput. Mech. 2022, 8, 370–378. [Google Scholar]
- Green, M.S.; Tobolsky, A.V. A New Approach to the Theory of Relaxing Polymeric Media. J. Chem. Phys. 1946, 14, 80–92. [Google Scholar] [CrossRef]
- Jenkins, R.D. The Fundamental Thickening Mechanism of Associative Polymers in Latex Systems: A Rheological Study; Lehigh University: Bethlehem, PA, USA, 1990. [Google Scholar]
- Jenkins, R.D.; Silebi, C.A.; El-Aasser, M.S. Steady-Shear and Linear-Viscoelastic Material Properties of Model Associative Polymer Solutions. In Polymers as Rheology Modifiers; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1991; Volume 462, pp. 222–233. ISBN 9780841220096. [Google Scholar]
- Tanaka, F.; Edwards, S.F. Viscoelastic Properties of Physically Crosslinked Networks: Part 2. Dynamic Mechanical Moduli. J. Non-Newtonian Fluid Mech. 1992, 43, 273–288. [Google Scholar] [CrossRef]
- Tanaka, F.; Edwards, S.F. Viscoelastic Properties of Physically Crosslinked Networks. 1. Transient Network Theory. Macromolecules 1992, 25, 1516–1523. [Google Scholar] [CrossRef]
- Tanaka, F. Theoretical Study of Molecular Association and Thermoreversible Gelation in Polymers. Polym. J. 2002, 34, 479–509. [Google Scholar] [CrossRef]
- Meng, F.; Saed, M.O.; Terentjev, E.M. Rheology of Vitrimers. Nat. Commun. 2022, 13, 5753. [Google Scholar] [CrossRef]
- Meng, F.; Saed, M.O.; Terentjev, E.M. Elasticity and Relaxation in Full and Partial Vitrimer Networks. Macromolecules 2019, 52, 7423–7429. [Google Scholar] [CrossRef]
- Meng, F.; Pritchard, R.H.; Terentjev, E.M. Stress Relaxation, Dynamics, and Plasticity of Transient Polymer Networks. Macromolecules 2016, 49, 2843–2852. [Google Scholar] [CrossRef]
- Meng, F.; Terentjev, E.M. Transient Network at Large Deformations: Elastic–Plastic Transition and Necking Instability. Polymers 2016, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Janeschitz-Kriegl, H. Polymer Melt Rheology and Flow Birefringence; Springer: Berlin/Heidelberg, Germany, 1983. [Google Scholar]
- Riande, E.; Saiz, E. Dipole Moments and Birefringence of Polymers; Prentice Hall: Hoboken, NJ, USA, 1992. [Google Scholar]
- Doi, M.; Edwards, S.F.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: London, UK, 1988; ISBN 9780198520337. [Google Scholar]
- Watanabe, H. Viscoelasticity and Dynamics of Entangled Polymers. Prog. Polym. Sci. 1999, 24, 1253–1403. [Google Scholar] [CrossRef]
- Shikata, T.; Pearson, D.S. Viscoelastic Behavior of Concentrated Spherical Suspensions. J. Rheol. 1994, 38, 601–616. [Google Scholar] [CrossRef]
- Parada, G.A.; Zhao, X. Ideal Reversible Polymer Networks. Soft Matter 2018, 14, 5186–5196. [Google Scholar] [CrossRef] [PubMed]
- Leibler, L.; Rubinstein, M.; Colby, R.H. Dynamics of Reversible Networks. Macromolecules 1991, 24, 4701–4707. [Google Scholar] [CrossRef]
- Katashima, T. Molecular Understanding of Viscoelasticity in Transient Polymer Networks Based on Multiple Methods. Nihon Reoroji Gakkaishi 2022, 50, 51–56. [Google Scholar] [CrossRef]
- Katashima, T. Rheological Studies on Polymer Networks with Static and Dynamic Crosslinks. Polym. J. 2021, 53, 1073–1082. [Google Scholar] [CrossRef]
- Annable, T.; Buscall, R.; Ettelaie, R.; Whittlestone, D. The Rheology of Solutions of Associating Polymers: Comparison of Experimental Behavior with Transient Network Theory. J. Rheol. 1993, 37, 695–726. [Google Scholar] [CrossRef]
- Suzuki, S.; Uneyama, T.; Inoue, T.; Watanabe, H. Rheology of Aqueous Solution of Hydrophobically Modified Ethoxylated Urethane (HEUR) with Fluorescent Probes at Chain Ends: Thinning Mechanism. Nihon Reoroji Gakkaishi 2012, 40, 31–36. [Google Scholar] [CrossRef]
- Tripathi, A.; Tam, K.C.; McKinley, G.H. Rheology and Dynamics of Associative Polymers in Shear and Extension: Theory and Experiments. Macromolecules 2006, 39, 1981–1999. [Google Scholar] [CrossRef]
- Le Meins, J.-F.; Tassin, J.-F. Shear-Induced Phase Separation in an Associating Polymer Solution. Macromolecules 2001, 34, 2641–2647. [Google Scholar] [CrossRef]
- Suzuki, S.; Uneyama, T.; Inoue, T.; Watanabe, H. Nonlinear Rheology of Telechelic Associative Polymer Networks: Shear Thickening and Thinning Behavior of Hydrophobically Modified Ethoxylated Urethane (HEUR) in Aqueous Solution. Macromolecules 2012, 45, 888–898. [Google Scholar] [CrossRef]
- Chiba, T.; Katashima, T.; Urakawa, O.; Inoue, T. Rheological Test for the Homogeneity of Aqueous Blends of Associative Polymer Network and Entangled Linear Polymer. Nihon Reoroji Gakkaishi 2020, 48, 49–54. [Google Scholar] [CrossRef]
- Katashima, T.; Kudo, R.; Naito, M.; Nagatoishi, S.; Miyata, K.; Chung, U.; Tsumoto, K.; Sakai, T. Experimental Comparison of Bond Lifetime and Viscoelastic Relaxation in Transient Networks with Well-Controlled Structures. ACS Macro Lett. 2022, 11, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Annable, T.; Ettelaie, R. Thermodynamics of Phase Separation in Mixtures of Associating Polymers and Homopolymers in Solution. Macromolecules 1994, 27, 5616–5622. [Google Scholar] [CrossRef]
- Annable, T.; Buscall, R.; Ettelaie, R. Network Formation and Its Consequences for the Physical Behaviour of Associating Polymers in Solution. Colloids Surf. A Physicochem. Eng. Asp. 1996, 112, 97–116. [Google Scholar] [CrossRef]
- Shabbir, A.; Huang, Q.; Chen, Q.; Colby, R.H.; Alvarez, N.J.; Hassager, O. Brittle Fracture in Associative Polymers: The Case of Ionomer Melts. Soft Matter 2016, 12, 7606–7612. [Google Scholar] [CrossRef]
- Uneyama, T.; Suzuki, S.; Watanabe, H. Concentration Dependence of Rheological Properties of Telechelic Associative Polymer Solutions. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2012, 86, 031802. [Google Scholar] [CrossRef]
- Winnik, M.A.; Yekta, A. Associative Polymers in Aqueous Solution. Curr. Opin. Colloid Interface Sci. 1997, 2, 424–436. [Google Scholar] [CrossRef]
- Dehara, K.; Yoshizaki, T.; Yamakawa, H. Translational Diffusion Coefficient of Oligo- and Poly(Methyl Methacrylates) in Dilute Solutions. Macromolecules 1993, 26, 5137–5142. [Google Scholar] [CrossRef]
- Phalakornkul, J.K.; Gast, A.P.; Pecora, R. Rotational and Translational Dynamics of Rodlike Polymers: A Combined Transient Electric Birefringence and Dynamic Light Scattering Study. Macromolecules 1999, 32, 3122–3135. [Google Scholar] [CrossRef]
- Nemoto, N.; Makita, Y.; Tsunashima, Y.; Kurata, M. Dynamic Light Scattering Studies of Polymer Solutions. 3. Translational Diffusion and Internal Motion of High Molecular Weight Polystyrenes in Benzene at Infinite Dilution. Macromolecules 1984, 17, 425–430. [Google Scholar] [CrossRef]
- Shokeen, N.; Issa, C.; Mukhopadhyay, A. Comparison of Nanoparticle Diffusion Using Fluorescence Correlation Spectroscopy and Differential Dynamic Microscopy within Concentrated Polymer Solutions. Appl. Phys. Lett. 2017, 111, 263703. [Google Scholar] [CrossRef]
- Alam, S.; Mukhopadhyay, A. Translational and Rotational Diffusions of Nanorods within Semidilute and Entangled Polymer Solutions. Macromolecules 2014, 47, 6919–6924. [Google Scholar] [CrossRef]
- Cush, R.; Dorman, D.; Russo, P.S. Rotational and Translational Diffusion of Tobacco Mosaic Virus in Extended and Globular Polymer Solutions. Macromolecules 2004, 37, 9577–9584. [Google Scholar] [CrossRef]
- Daivis, P.; Snook, I.; Van Megen, W.; Preston, B.N.; Comper, W.D. Dynamic Light Scattering Measurements of Diffusion in Polymer-Polymer-Solvent Systems. Macromolecules 1984, 17, 2376–2380. [Google Scholar] [CrossRef]
- Burchard, W.; Richtering, W. Dynamic Light Scattering from Polymer Solutions. In Relaxation in Polymers; Progress in Colloid & Polymer Science; Steinkopff: Hannover, Germany, 1989; Volume 80, pp. 151–163. [Google Scholar]
- Shibayama, M. Spatial Inhomogeneity and Dynamic Fluctuations of Polymer Gels. Macromol. Chem. Phys. 1998, 199, 1–30. [Google Scholar] [CrossRef]
- Nicolai, T.; Brown, W. Cooperative Diffusion of Concentrated Polymer Solutions: A Static and Dynamic Light Scattering Study of Polystyrene in DOP. Macromolecules 1996, 29, 1698–1704. [Google Scholar] [CrossRef]
- Amis, E.J.; Han, C.C. Cooperative and Self-Diffusion of Polymers in Semidilute Solutions by Dynamic Light Scattering. Polymer 1982, 23, 1403–1406. [Google Scholar] [CrossRef]
- Fujiyabu, T.; Sakai, T.; Kudo, R.; Yoshikawa, Y.; Katashima, T.; Chung, U.; Sakumichi, N. Temperature Dependence of Polymer Network Diffusion. Phys. Rev. Lett. 2021, 127, 237801. [Google Scholar] [CrossRef] [PubMed]
- Kureha, T.; Minato, H.; Suzuki, D.; Urayama, K.; Shibayama, M. Concentration Dependence of the Dynamics of Microgel Suspensions Investigated by Dynamic Light Scattering. Soft Matter 2019, 15, 5390–5399. [Google Scholar] [CrossRef] [PubMed]
- Meerwall, E.D. Self-Diffusion in Polymer Systems, Measured with Field-Gradient Spin Echo NMR Methods. In Spectroscopy: NMR, Fluorescence, FT-IR; Fortschritte der Hochpolymeren-Forschung [Advances in Polymer Science]; Springer: Berlin/Heidelberg, Germany, 1984; pp. 1–29. ISBN 9783540125914. [Google Scholar]
- Kruteva, M.; Allgaier, J.; Richter, D. Direct Observation of Two Distinct Diffusive Modes for Polymer Rings in Linear Polymer Matrices by Pulsed Field Gradient (PFG) NMR. Macromolecules 2017, 50, 9482–9493. [Google Scholar] [CrossRef]
- Fu, L.; Pacheco, C.; Prud’homme, R.K. Translational and Rotational Diffusion of Globular Proteins in Concentrated Polymer Networks. Soft Mater. 2009, 7, 213–231. [Google Scholar] [CrossRef]
- Pagès, G.; Gilard, V.; Martino, R.; Malet-Martino, M. Pulsed-Field Gradient Nuclear Magnetic Resonance Measurements (PFG NMR) for Diffusion Ordered Spectroscopy (DOSY) Mapping. Analyst 2017, 142, 3771–3796. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, J.; Jarvet, J.; Damberg, P.; Gräslund, A. Translational Diffusion Measured by PFG-NMR on Full Length and Fragments of the Alzheimer Aβ(1–40) Peptide. Determination of Hydrodynamic Radii of Random Coil Peptides of Varying Length. Magn. Reson. Chem. 2002, 40, S89–S97. [Google Scholar] [CrossRef]
- Guo, X.; Wetzel, K.S.; Solleder, S.C.; Spann, S.; Meier, M.A.R.; Wilhelm, M.; Luy, B.; Guthausen, G. 1H PFG-NMR Diffusion Study on a Sequence-defined Macromolecule: Confirming Monodispersity. Macromol. Chem. Phys. 2019, 220, 1900155. [Google Scholar] [CrossRef]
- Kärger, J.; Stallmach, F. PFG NMR Studies of Anomalous Diffusion. In Diffusion in Condensed Matter: Methods, Materials, Models; Heitjans, P., Kärger, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 417–459. ISBN 9783540309703. [Google Scholar]
- Skirda, V.D. The Features of PFG NMR Technique and Some Methodical Aspects of Its Application. In Magnetic Resonance in Colloid and Interface Science; Fraissard, J., Lapina, O., Eds.; Springer: Dordrecht, The Netherlands, 2002; pp. 245–254. ISBN 9789401005340. [Google Scholar]
- Sprague, B.L.; McNally, J.G. FRAP Analysis of Binding: Proper and Fitting. Trends Cell Biol. 2005, 15, 84–91. [Google Scholar] [CrossRef]
- Moud, A.A. Fluorescence Recovery after Photobleaching in Colloidal Science: Introduction and Application. ACS Biomater. Sci. Eng. 2022, 8, 1028–1048. [Google Scholar] [CrossRef]
- Sustr, D.; Hlaváček, A.; Duschl, C.; Volodkin, D. Multi-Fractional Analysis of Molecular Diffusion in Polymer Multilayers by FRAP: A New Simulation-Based Approach. J. Phys. Chem. B 2018, 122, 1323–1333. [Google Scholar] [CrossRef]
- Hauser, G.I.; Seiffert, S.; Oppermann, W. Systematic Evaluation of FRAP Experiments Performed in a Confocal Laser Scanning Microscope--Part II: Multiple Diffusion Processes. J. Microsc. 2008, 230, 353–362. [Google Scholar] [CrossRef]
- Seiffert, S.; Oppermann, W. Systematic Evaluation of FRAP Experiments Performed in a Confocal Laser Scanning Microscope. J. Microsc. 2005, 220, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Ferry, J.D. Viscoelastic Properties of Polymers; John Wiley & Sons: New York, NY, USA, 1980; ISBN 9780471048947. [Google Scholar]
- Ohnishi, M.; Katashima, T.; Nakahata, M.; Urakawa, O. Relationships between Diffusion and Viscoelasticity of Associative Polymer Networks. Nihon Reoroji Gakkaishi 2019, 47, 133–142. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The Physics of Rubber Elasticity; OUP: Oxford, UK, 1975; ISBN 9780191523304. [Google Scholar]
- Yamamoto, R.; Onuki, A. Dynamics of Highly Supercooled Liquids: Heterogeneity, Rheology, and Diffusion. Phys. Rev. E 1998, 58, 3515–3529. [Google Scholar] [CrossRef]
- Sillescu, H. Heterogeneity at the Glass Transition: A Review. J. Non-Cryst. Solids 1999, 243, 81–108. [Google Scholar] [CrossRef]
- Binder, K.; Kob, W. Glassy Materials and Disordered Solids. Available online: https://www.worldscientific.com/worldscibooks/10.1142/7300 (accessed on 18 November 2023).
- Katashima, T.; Kobayashi, R.; Ishikawa, S.; Naito, M.; Miyata, K.; Chung, U.; Sakai, T. Decoupling between Translational Diffusion and Viscoelasticity in Transient Networks with Controlled Network Connectivity. Gels 2022, 8, 830. [Google Scholar] [CrossRef]
- Kato, M.; Ishikawa, S.; Shen, Q.; Du, Z.; Katashima, T.; Naito, M.; Numahata, T.; Okazaki, M.; Sakai, T.; Kurita, M. In Situ-Formable, Dynamic Crosslinked Poly (Ethylene Glycol) Carrier for Localized Adeno-Associated Virus Infection and Reduced off-Target Effects. Commun. Biol. 2023, 6, 508. [Google Scholar] [CrossRef] [PubMed]
- Katashima, T.; Kudo, R.; Onishi, R.; Naito, M.; Nagatoishi, S.; Miyata, K.; Tsumoto, K.; Chung, U.; Sakai, T. Effects of Network Connectivity on Viscoelastic Relaxation in Transient Networks Using Experimental Approach. Front. Soft. Matter 2022, 2, 1059156. [Google Scholar] [CrossRef]
- Ge, S.; Tress, M.; Xing, K.; Cao, P.-F.; Saito, T.; Sokolov, A.P. Viscoelasticity in Associating Oligomers and Polymers: Experimental Test of the Bond Lifetime Renormalization Model. Soft Matter 2020, 16, 390–401. [Google Scholar] [CrossRef]
- Shabbir, A.; Javakhishvili, I.; Cerveny, S.; Hvilsted, S.; Skov, A.L.; Hassager, O.; Alvarez, N.J. Linear Viscoelastic and Dielectric Relaxation Response of Unentangled UPy-Based Supramolecular Networks. Macromolecules 2016, 49, 3899–3910. [Google Scholar] [CrossRef]
- Marco-Dufort, B.; Tibbitt, M.W. Design of Moldable Hydrogels for Biomedical Applications Using Dynamic Covalent Boronic Esters. Mater. Today Chem. 2019, 12, 16–33. [Google Scholar] [CrossRef]
- Sheridan, R.J.; Bowman, C.N. A Simple Relationship Relating Linear Viscoelastic Properties and Chemical Structure in a Model Diels–Alder Polymer Network. Macromolecules 2012, 45, 7634–7641. [Google Scholar] [CrossRef]
- Singh, P. SPR Biosensors: Historical Perspectives and Current Challenges. Sens. Actuators B Chem. 2016, 229, 110–130. [Google Scholar] [CrossRef]
- Benesi, H.A.; Hildebrand, J.H. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Shulyak, I.V.; Grushova, E.I.; Semenchenko, A.M. Rheological Properties of Aqueous Solutions of Polyethylene Glycols with Various Molecular Weights. Russ. J. Phys. Chem. A 2011, 85, 419–422. [Google Scholar] [CrossRef]
- Semenov, A.N.; Rubinstein, M. Thermoreversible Gelation in Solutions of Associative Polymers. 1. Statics. Macromolecules 1998, 31, 1373–1385. [Google Scholar] [CrossRef]
- Chen, Q.; Huang, C.; Weiss, R.A.; Colby, R.H. Viscoelasticity of Reversible Gelation for Ionomers. Macromolecules 2015, 48, 1221–1230. [Google Scholar] [CrossRef]
- Sakai, T.; Katashima, T.; Matsushita, T.; Chung, U. Sol-Gel Transition Behavior near Critical Concentration and Connectivity. Polym. J. 2016, 48, 629–634. [Google Scholar] [CrossRef]
- Katashima, T.; Sakurai, H.; Chung, U.; Sakai, T. Dilution Effect on the Cluster Growth near the Gelation Threshold. Nihon Reoroji Gakkaishi 2019, 47, 61–66. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michida, S.; Chung, U.-i.; Katashima, T. Probing the Molecular Mechanism of Viscoelastic Relaxation in Transient Networks. Gels 2023, 9, 945. https://doi.org/10.3390/gels9120945
Michida S, Chung U-i, Katashima T. Probing the Molecular Mechanism of Viscoelastic Relaxation in Transient Networks. Gels. 2023; 9(12):945. https://doi.org/10.3390/gels9120945
Chicago/Turabian StyleMichida, Shota, Ung-il Chung, and Takuya Katashima. 2023. "Probing the Molecular Mechanism of Viscoelastic Relaxation in Transient Networks" Gels 9, no. 12: 945. https://doi.org/10.3390/gels9120945