
Para-Methoxybenzylidene Acetal-Protected D-Glucosamine Derivatives as pH-Responsive Gelators and Their Applications for Drug Delivery

Abstract
:1. Introduction
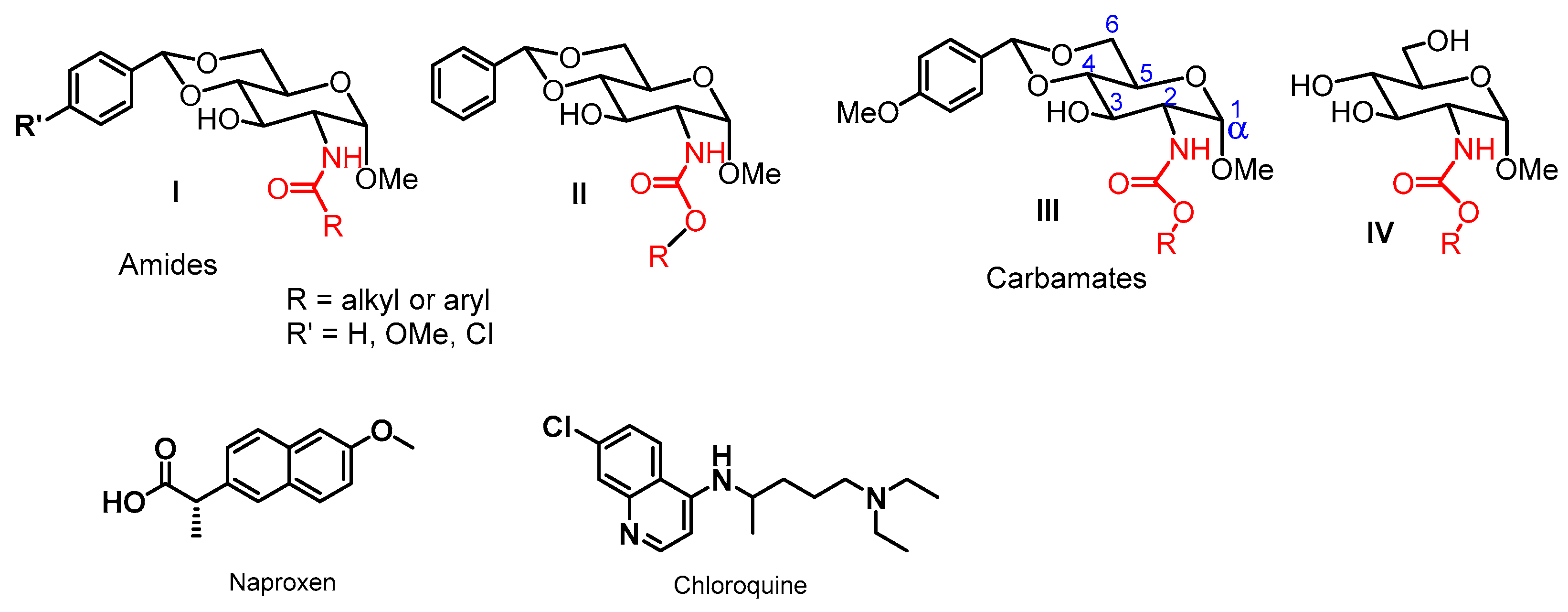
2. Results and Discussion
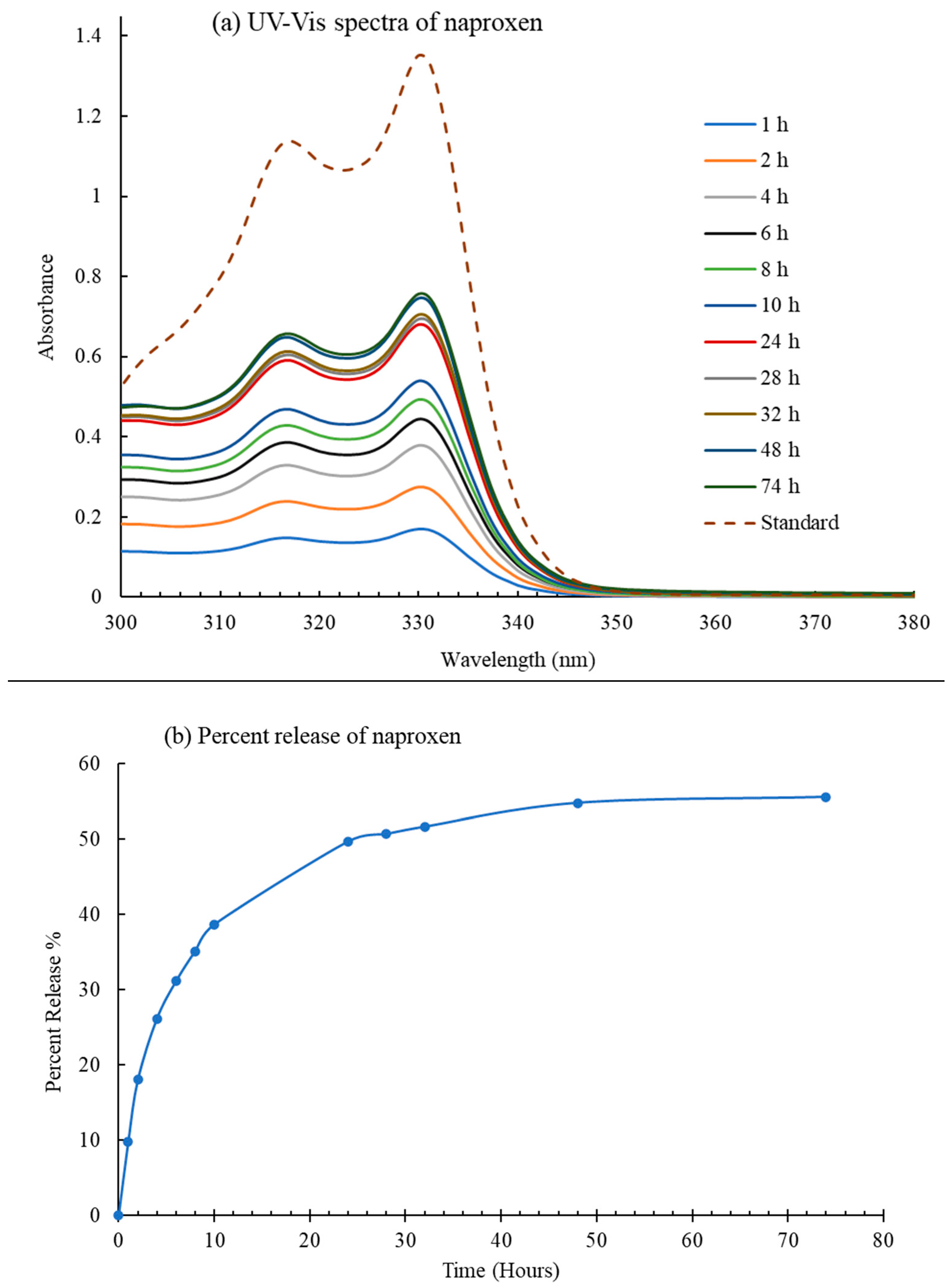
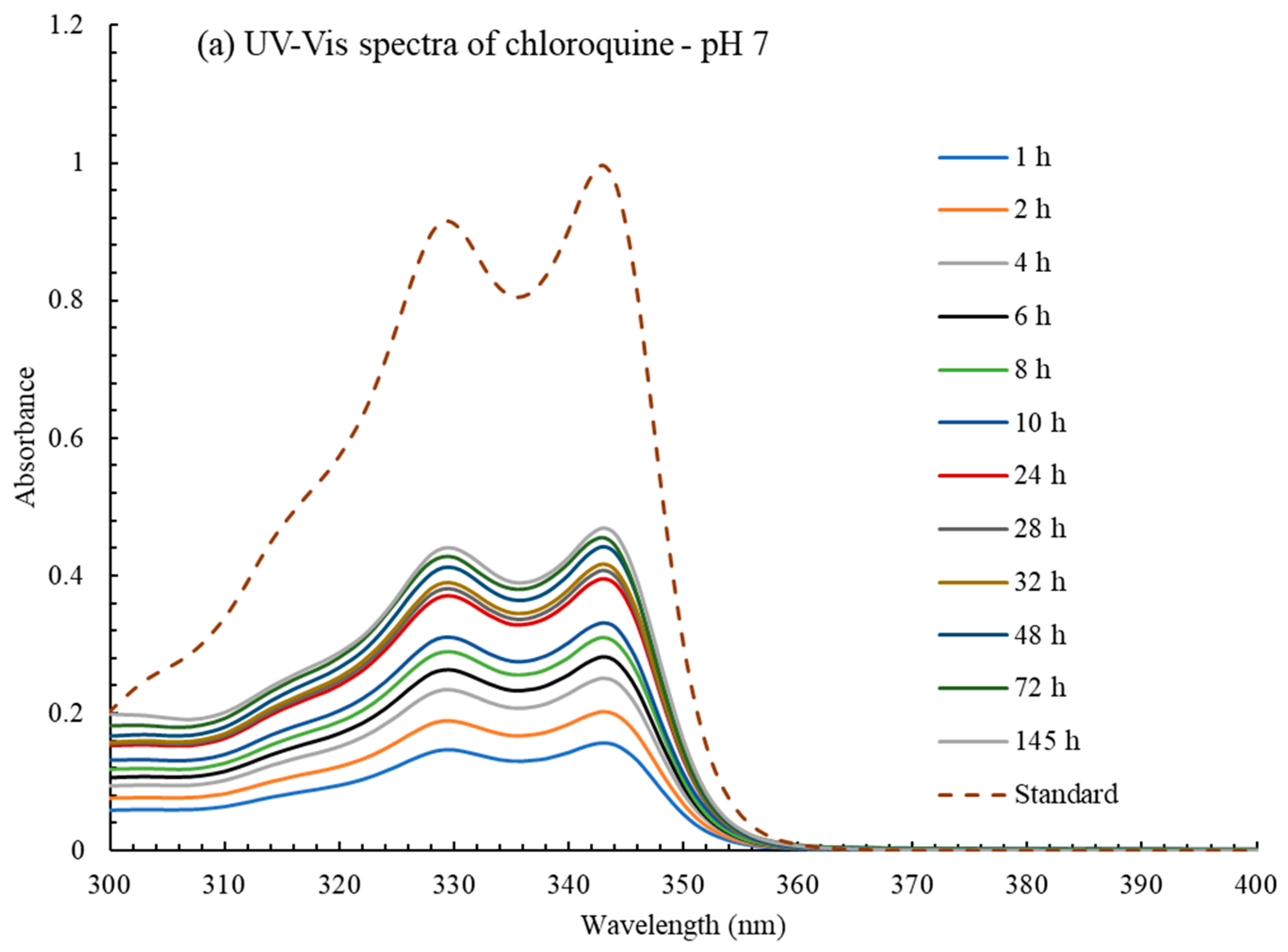
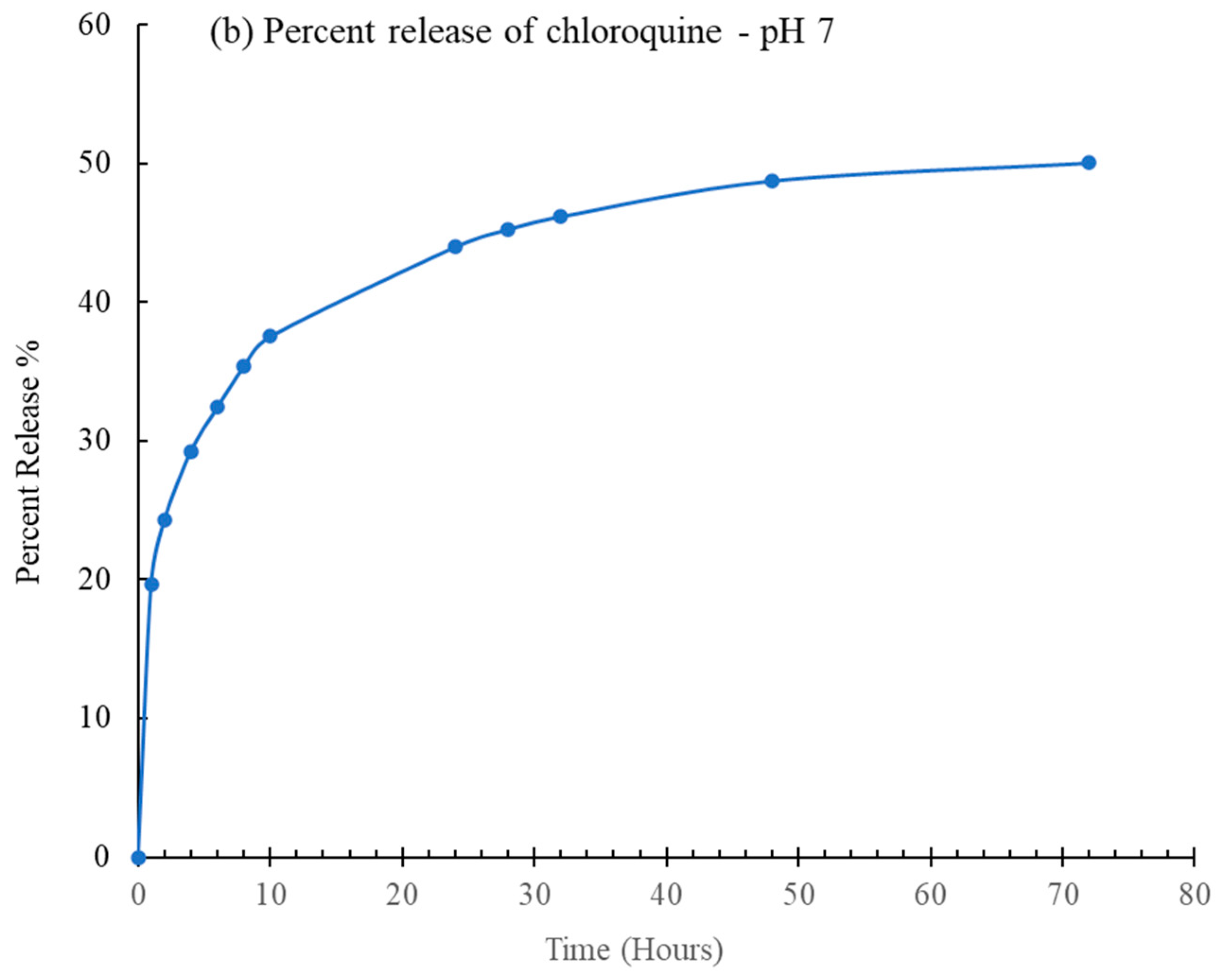
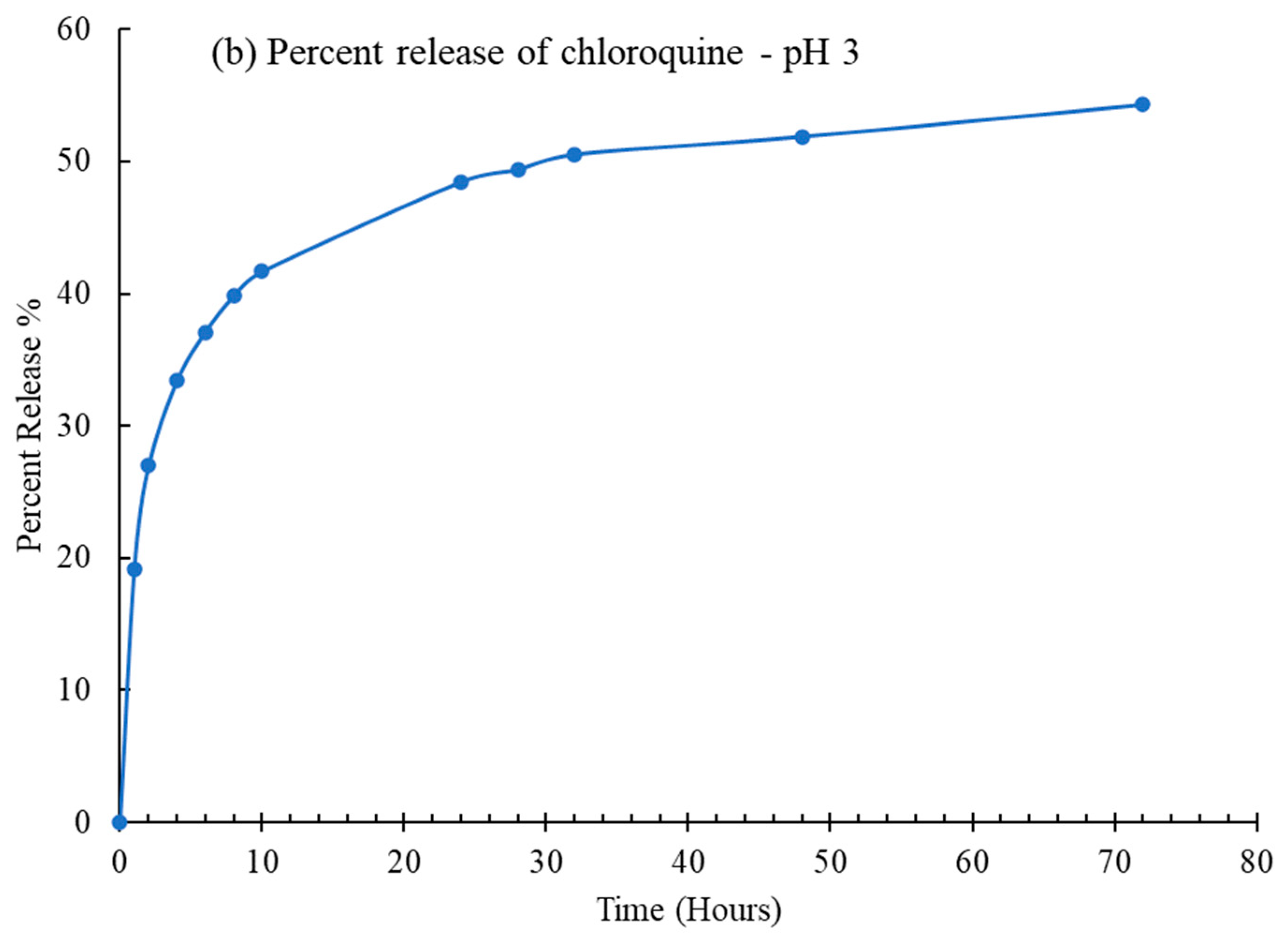
2.1. Drug Delivery Studies
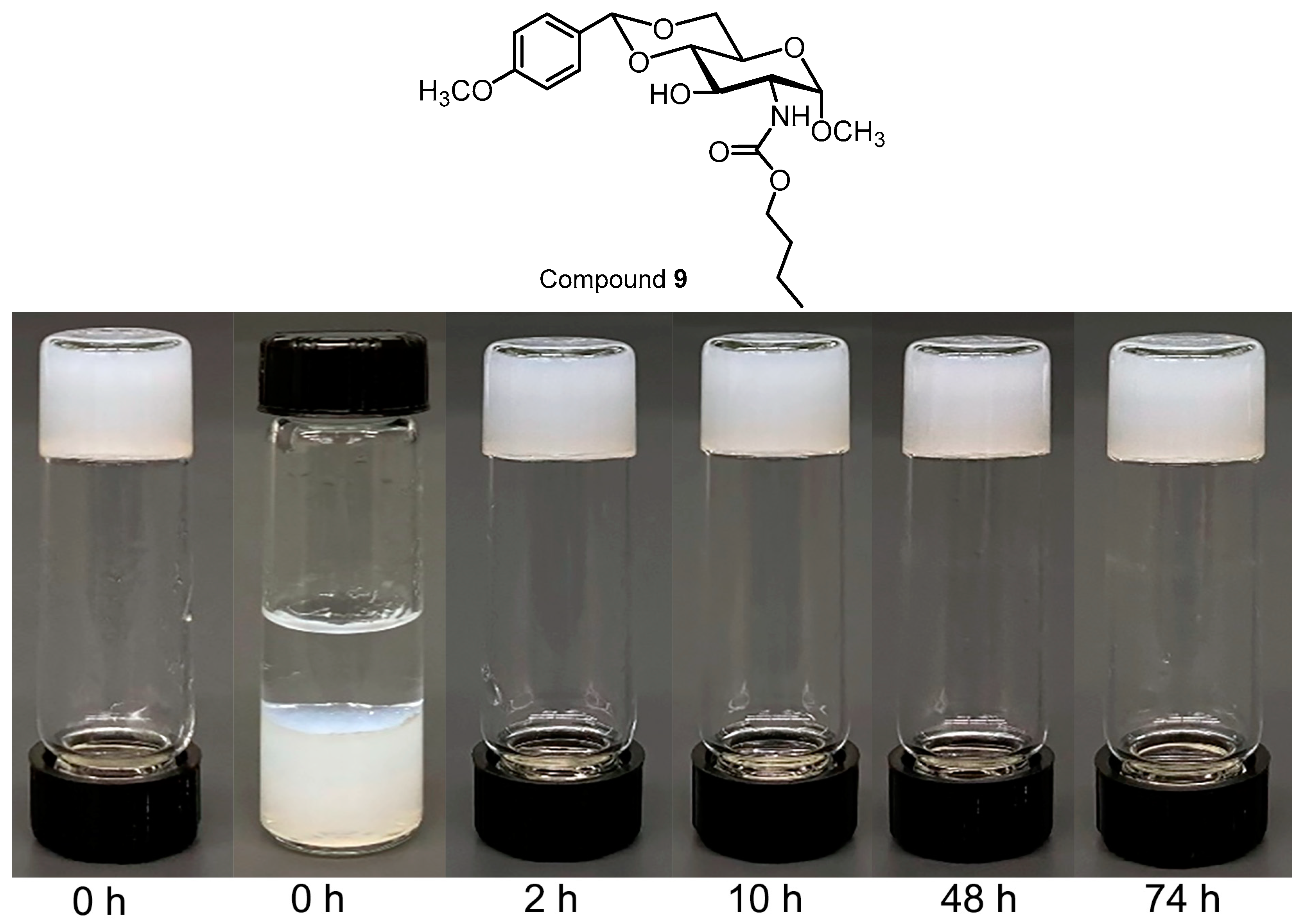
2.2. Acid-Triggering Gelation Studies
3. Conclusions
4. Materials and Methods
4.1. Naproxen Trapping and Release Studies
4.2. Chloroquine Trapping and Release Studies
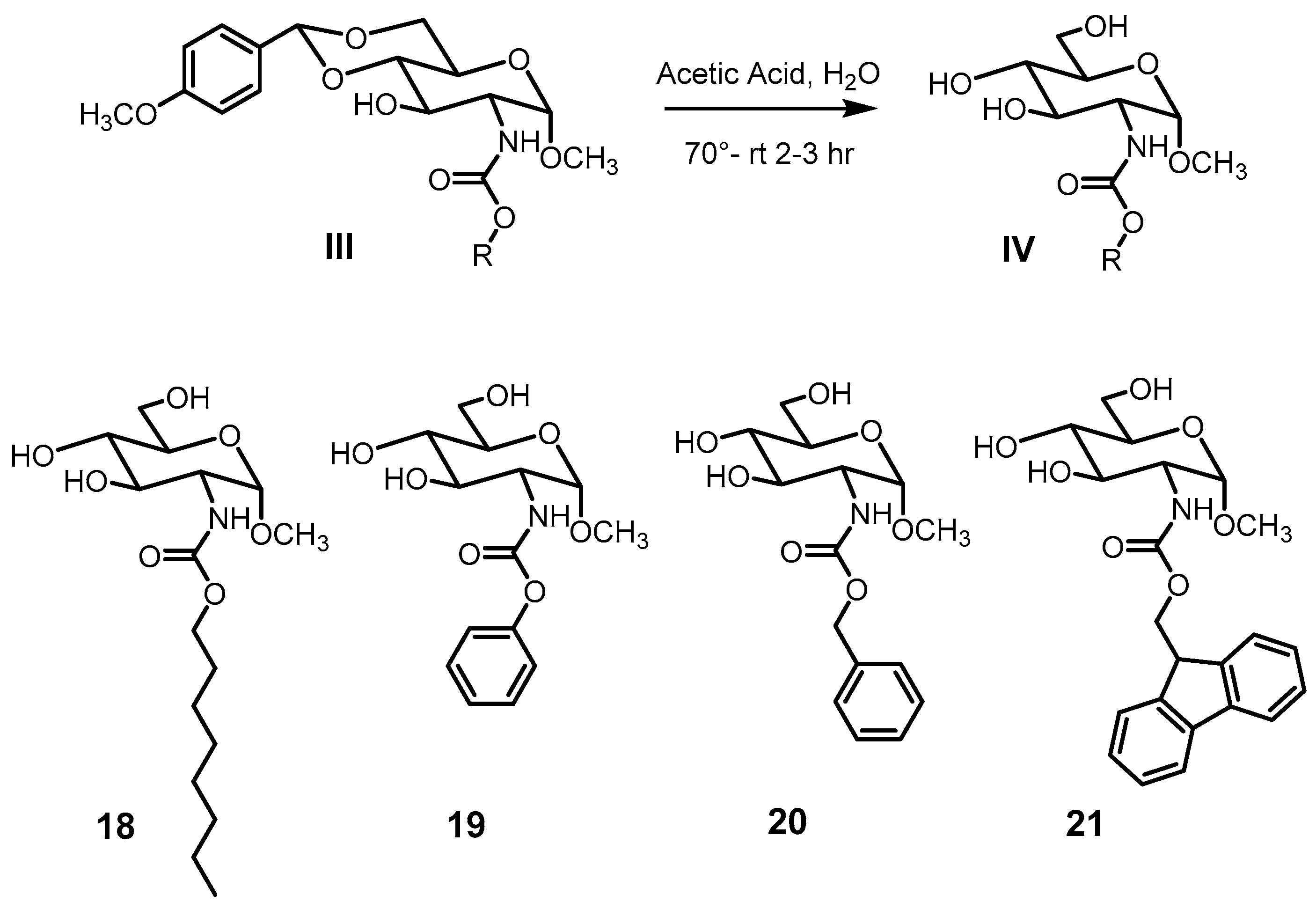
4.3. General Procedure for the Preparation of Carbamate Compounds 3–4
4.4. General Procedure for the Preparation of Carbamate Compounds 5–17
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vemula, P.K.; John, G. Crops: A green approach toward self-assembled soft materials. Acc. Chem. Res. 2008, 41, 769–782. [Google Scholar] [CrossRef]
- Datta, S.; Bhattacharya, S. Multifarious facets of sugar-derived molecular gels: Molecular features, mechanisms of self-assembly and emerging applications. Chem. Soc. Rev. 2015, 44, 5596–5637. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Shi, J.; Xu, B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.; Bietsch, J.; Bashaw, K.; Wang, G. Recently Developed Carbohydrate Based Gelators and Their Applications. Gels 2021, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Basu, N.; Chakraborty, A.; Ghosh, R. Carbohydrate derived organogelators and the corresponding functional gels developed in recent time. Gels 2018, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Vibhute, A.M.; Muvvala, V.; Sureshan, K.M. A Sugar-Based Gelator for Marine Oil-Spill Recovery. Angew. Chem. Int. Ed. 2016, 55, 7782–7785. [Google Scholar] [CrossRef]
- Bhuniya, S.; Kim, B.H. An insulin-sensing sugar-based fluorescent hydrogel. Chem. Commun. 2006, 17, 1842–1844. [Google Scholar] [CrossRef] [PubMed]
- Hemamalini, A.; Das, T.M. Design and synthesis of sugar-triazole low molecular weight gels as mercury ion sensor. New J. Chem. 2013, 37, 2419–2425. [Google Scholar] [CrossRef]
- Khayat, Z.; Zali-Boeini, H. Novel sugar-based azo dyes as multistimuli responsive supramolecular gelators and chemosensors. Dyes Pigm. 2018, 159, 337–344. [Google Scholar] [CrossRef]
- Wang, G.; Wang, D.; Bietsch, J.; Chen, A.; Sharma, P. Synthesis of Dendritic Glycoclusters and Their Applications for Supramolecular Gelation and Catalysis. J. Org. Chem. 2020, 85, 16136–16156. [Google Scholar] [CrossRef]
- Hawkins, K.; Patterson, A.K.; Clarke, P.A.; Smith, D.K. Catalytic Gels for a Prebiotically Relevant Asymmetric Aldol Reaction in Water: From Organocatalyst Design to Hydrogel Discovery and Back Again. J. Am. Chem. Soc. 2020, 142, 4379–4389. [Google Scholar] [CrossRef]
- Diaz Diaz, D.; Kuhbeck, D.; Koopmans, R.J. Stimuli-responsive gels as reaction vessels and reusable catalysts. Chem. Soc. Rev. 2011, 40, 427–448. [Google Scholar] [CrossRef]
- Dou, X.-Q.; Yang, X.-M.; Li, P.; Zhang, Z.-G.; Schönherr, H.; Zhang, D.; Feng, C.-L. Novel pH responsive hydrogels for controlled cell adhesion and triggered surface detachment. Soft Matter 2012, 8, 9539. [Google Scholar] [CrossRef]
- Giraud, T.; Hoschtettler, P.; Pickaert, G.; Averlant-Petit, M.-C.; Stefan, L. Emerging low-molecular weight nucleopeptide-based hydrogels: State of the art, applications, challenges and perspectives. Nanoscale 2022, 14, 4908–4921. [Google Scholar] [CrossRef] [PubMed]
- Prathap, A.; Sureshan, K.M. Sugar-Based Organogelators for Various Applications. Langmuir 2019, 35, 6005–6014. [Google Scholar] [CrossRef]
- Pires, R.A.; Abul-Haija, Y.M.; Costa, D.S.; Novoa-Carballal, R.; Reis, R.L.; Ulijn, R.V.; Pashkuleva, I. Controlling Cancer Cell Fate Using Localized Biocatalytic Self-Assembly of an Aromatic Carbohydrate Amphiphile. J. Am. Chem. Soc. 2015, 137, 576–579. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wu, S.; Liu, D.; Chi, C.; Zhang, W.; Ma, M.; Lai, L.; Dong, S. Glycopeptide Self-Assembly Modulated by Glycan Stereochemistry through Glycan-Aromatic Interactions. J. Am. Chem. Soc. 2020, 142, 17015–17023. [Google Scholar] [CrossRef]
- Brito, A.; Kassem, S.; Reis, R.L.; Ulijn, R.V.; Pires, R.A.; Pashkuleva, I. Carbohydrate amphiphiles for supramolecular biomaterials: Design, self-assembly, and applications. Chem 2021, 7, 2943–2964. [Google Scholar] [CrossRef]
- Zhou, J.; Li, J.; Du, X.; Xu, B. Supramolecular biofunctional materials. Biomaterials 2017, 129, 1–27. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Xu, B. Supramolecular Hydrogels Made of Basic Biological Building Blocks. Chem. Asian J. 2014, 9, 1446–1472. [Google Scholar] [CrossRef]
- Chivers, P.R.A.; Smith, D.K. Spatially-resolved soft materials for controlled release–hybrid hydrogels combining a robust photo-activated polymer gel with an interactive supramolecular gel. Chem. Sci. 2017, 8, 7218–7227. [Google Scholar] [CrossRef] [PubMed]
- Kapros, A.; Balazs, A.; Harmat, V.; Halo, A.; Budai, L.; Pinter, I.; Menyhard, D.K.; Perczel, A. Configuration-Controlled Crystal and/or Gel Formation of Protected D-Glucosamines Supported by Promiscuous Interaction Surfaces and a Conformationally Heterogeneous Solution State. Chem. Eur. J. 2020, 26, 11643–11655. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Cheuk, S.; Yang, H.; Goyal, N.; Reddy, P.V.N.; Hopkinson, B. Synthesis and Characterization of Monosaccharide-Derived Carbamates as Low-Molecular-Weight Gelators. Langmuir 2009, 25, 8696–8705. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.; Mangunuru, H.P.R.; Parikh, B.; Shrestha, S.; Wang, G. Synthesis and characterization of pH responsive D-glucosamine based molecular gelators. Beilstein J. Org. Chem. 2014, 10, 3111–3121. [Google Scholar] [CrossRef] [PubMed]
- Bietsch, J.; Olson, M.; Wang, G. Fine-Tuning of Molecular Structures to Generate Carbohydrate Based Super Gelators and Their Applications for Drug Delivery and Dye Absorption. Gels 2021, 7, 134. [Google Scholar] [CrossRef]
- Goyal, N.; Cheuk, S.; Wang, G. Synthesis and characterization of D-glucosamine-derived low molecular weight gelators. Tetrahedron 2010, 66, 5962–5971. [Google Scholar] [CrossRef]
- Zhang, X.; Malhotra, S.; Molina, M.; Haag, R. Micro- and nanogels with labile crosslinks—From synthesis to biomedical applications. Chem. Soc. Rev. 2015, 44, 1948–1973. [Google Scholar] [CrossRef]
- Mayr, J.; Saldias, C.; Diaz Diaz, D. Release of small bioactive molecules from physical gels. Chem. Soc. Rev. 2018, 47, 1484–1515. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, X.; Wu, Y.; Zhang, J.; Zhu, Y.; Wang, J. A multifunctional supramolecular hydrogel: Preparation, properties and molecular assembly. Soft Matter 2018, 14, 566–573. [Google Scholar] [CrossRef]
- Vilaca, H.; Pereira, G.; Castro, T.G.; Hermenegildo, B.F.; Shi, J.; Faria, T.Q.; Micaelo, N.; Brito, R.M.M.; Xu, B.; Castanheira, E.M.S.; et al. New self-assembled supramolecular hydrogels based on dehydropeptides. J. Mater. Chem. B 2015, 3, 6355–6367. [Google Scholar] [CrossRef]
- Wang, D.; Chen, A.; Morris, J.; Wang, G. Stimuli-responsive gelators from carbamoyl sugar derivatives and their responses to metal ions and tetrabutylammonium salts. RSC Adv. 2020, 10, 40068–40083. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.K.; El-Qarra, L.H.; Smith, D.K. Chirality-directed hydrogel assembly and interactions with enantiomers of an active pharmaceutical ingredient. Chem. Commun. 2022, 58, 3941–3944. [Google Scholar] [CrossRef] [PubMed]
- Marti-Centelles, R.; Dolz-Perez, I.; De la O, J.; Ontoria-Oviedo, I.; Sepulveda, P.; Nebot, V.J.; Vicent, M.J.; Escuder, B. Two-Component Peptidic Molecular Gels for Topical Drug Delivery of Naproxen. ACS Appl. Bio Mater. 2021, 4, 935–944. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, X.; Gu, J.; Chang, D.; Zhang, J.; Chen, Z.; Ye, Y.; Wang, C.; Tao, W.; Zeng, X.; et al. Investigation and intervention of autophagy to guide cancer treatment with nanogels. Nanoscale 2017, 9, 150–163. [Google Scholar] [CrossRef]
- Movellan, J.; Urban, P.; Moles, E.; de la Fuente, J.M.; Sierra, T.; Serrano, J.L.; Fernandez-Busquets, X. Amphiphilic dendritic derivatives as nanocarriers for the targeted delivery of antimalarial drugs. Biomaterials 2014, 35, 7940–7950. [Google Scholar] [CrossRef]
- Brueckner, M.; Scheffler, K.; Reibetanz, U. Enhanced cytoplasmic release of drug delivery systems: Chloroquine as a multilayer and template constituent of layer-by-layer microcarriers. J. Mater. Chem. B 2018, 6, 5153–5163. [Google Scholar] [CrossRef] [PubMed]
- Sleightholm, R.; Yang, B.; Yu, F.; Xie, Y.; Oupicky, D. Chloroquine-Modified Hydroxyethyl Starch as a Polymeric Drug for Cancer Therapy. Biomacromolecules 2017, 18, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Brito, A.; Abul-Haija, Y.M.; da Costa, D.S.; Novoa-Carballal, R.; Reis, R.L.; Ulijn, R.V.; Pires, R.A.; Pashkuleva, I. Minimalistic supramolecular proteoglycan mimics by co-assembly of aromatic peptide and carbohydrate amphiphiles. Chem. Sci. 2019, 10, 2385–2390. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, H.; Yang, Z.; Xu, B. Supramolecular hydrogels respond to ligand-receptor interaction. J. Am. Chem. Soc. 2003, 125, 13680–13681. [Google Scholar] [CrossRef]
- Prakash, V.; Christian, Y.; Redkar, A.S.; Roy, A.; Anandalakshmi, R.; Ramakrishnan, V. Antibacterial hydrogels of aromatic tripeptides. Soft Matter 2022, 18, 6360–6371. [Google Scholar] [CrossRef]
- Coleman, R.S.; Dong, Y.; Carpenter, A.J. A convenient preparation of terminally differentiated, selectively protected six-carbon synthons from D-glucosamine. J. Org. Chem. 1992, 57, 3732–3735. [Google Scholar] [CrossRef]
- Hanessian, S.; Maianti, J.P.; Matias, R.D.; Feeney, L.A.; Armstrong, E.S. Hybrid Aminoglycoside Antibiotics via Tsuji Palladium-Catalyzed Allylic Deoxygenation. Org. Lett. 2011, 13, 6476–6479. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.C.; Carbain, B.; Beale, G.S.; Alhasan, S.F.; Reeves, H.L.; Baisch, U.; Newell, D.R.; Golding, B.T.; Griffin, R.J. Regioselective sulfamoylation at low temperature enables concise syntheses of putative small molecule inhibitors of sulfatases. Org. Biomol. Chem. 2015, 13, 5279–5284. [Google Scholar] [CrossRef]
- Ying, L.; Gervay-Hague, J. Synthesis of N-(fluoren-9-ylmethoxycarbonyl)glycopyranosylamine uronic acids. Carbohydr. Res. 2004, 339, 367–375. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Toluene | EtOH | i-PrOH | n-PrOH | n-BuOH | EG | Glycerol | EtOH:H2O 1:1 | EtOH:H2O 1:2 | DMSO:H2O 1:1 | DMSO:H2O 1:2 | H2O | |
| 5 | GO 4.0 | GO 10.0 | P | GO 10.0 | GO 10.0 | GO 20.0 | GO 20.0 | GO 5.0 | GO 5.0 | GO 6.7 | GO 5.0 | GO 5.0 |
| 6 | GC 6.7 | GO 6.7 | GO 6.7 | GO 10.0 | GO 10.0 | GT 6.7 | GC 6.7 | GO 3.3 | GO 6.7 | GO 6.7 | GO 20 | I |
| 7 | S | GO 6.7 | GO 10.0 | S | S | GO 6.7 | GO 20.0 | GO 2.2 | GO 1.8 | GO 4.0 | GO 3.3 | GO 1.5 |
| 8 | P | GO 6.7 | P | GO 10.0 | S | GO 10.0 | S | GO 6.7 | GO 5.0 | GO 6.7 | P | I |
| 9 | P | P | P | P | P | P | S | GO 5.0 | GO 2.2 | GO 4.0 | GO 5.0 | GT 0.9 |
| 10 | S | S | S | S | S | S | GC 10.0 | P | P | P | P | I |
| 11 | P | GO 10.0 | GO 20.0 | GO 20.0 | GO 20.0 | S | UG | GO 6.7 | P | P | P | I |
| 12 | P | GO 4.0 | GO 5.0 | GO 10.0 | GO 10.0 | GO 20.0 | GO 10.0 | GO 5.0 | GO 5.0 | GO 5.0 | GO 5.0 | P |
| 13 | S | P | P | S | S | P | S | I | I | P | P | I |
| 14 | S | S | S | S | S | GO 20.0 | S | P | P | P | P | I |
| 15 | GO 5.0 | P | GO 10.0 | GO 10.0 | GO 20.0 | GO 10.0 | GO 10.0 | GO 2.2 | P | GT 2.2 | GO 4.0 | I |
| 16 | P | S | S | S | S | S | S | I | I | S | P | I |
| 17 | GT 10.0 | GT 2.8 | GT 5.0 | GT 4.0 | GT 5.0 | GT 5.0 | GT 20.0 | GT 0.8 | I | GT 1.7 | GT 1.0 | I |
| 18 | GT 2.9 | S | S | S | S | S | S | P | GO 10.0 | GO 10.0 | GO 10.0 | GO10.0 |
| 19 | GO 6.7 | S | S | S | S | S | S | S | S | S | S | P |
| 20 | Gc2.9 | P | S | S | S | S | S | P | P | S | S | I |
| 21 | GO 6.7 | GO 10.0 | GO 10.0 | GO 20.0 | GO 10.0 | GO 10.0 | GC 5.0 | GO 5.0 | GC 1.0 | GO 10.0 | GT 4.0 | GT 0.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bietsch, J.; Baker, L.; Duffney, A.; Mao, A.; Foutz, M.; Ackermann, C.; Wang, G. Para-Methoxybenzylidene Acetal-Protected D-Glucosamine Derivatives as pH-Responsive Gelators and Their Applications for Drug Delivery. Gels 2023, 9, 445. https://doi.org/10.3390/gels9060445
Bietsch J, Baker L, Duffney A, Mao A, Foutz M, Ackermann C, Wang G. Para-Methoxybenzylidene Acetal-Protected D-Glucosamine Derivatives as pH-Responsive Gelators and Their Applications for Drug Delivery. Gels. 2023; 9(6):445. https://doi.org/10.3390/gels9060445
Chicago/Turabian StyleBietsch, Jonathan, Logan Baker, Anna Duffney, Alice Mao, Mary Foutz, Cheandri Ackermann, and Guijun Wang. 2023. "Para-Methoxybenzylidene Acetal-Protected D-Glucosamine Derivatives as pH-Responsive Gelators and Their Applications for Drug Delivery" Gels 9, no. 6: 445. https://doi.org/10.3390/gels9060445