3.1. Characterization of AOA and Modification of AOA by Polymers
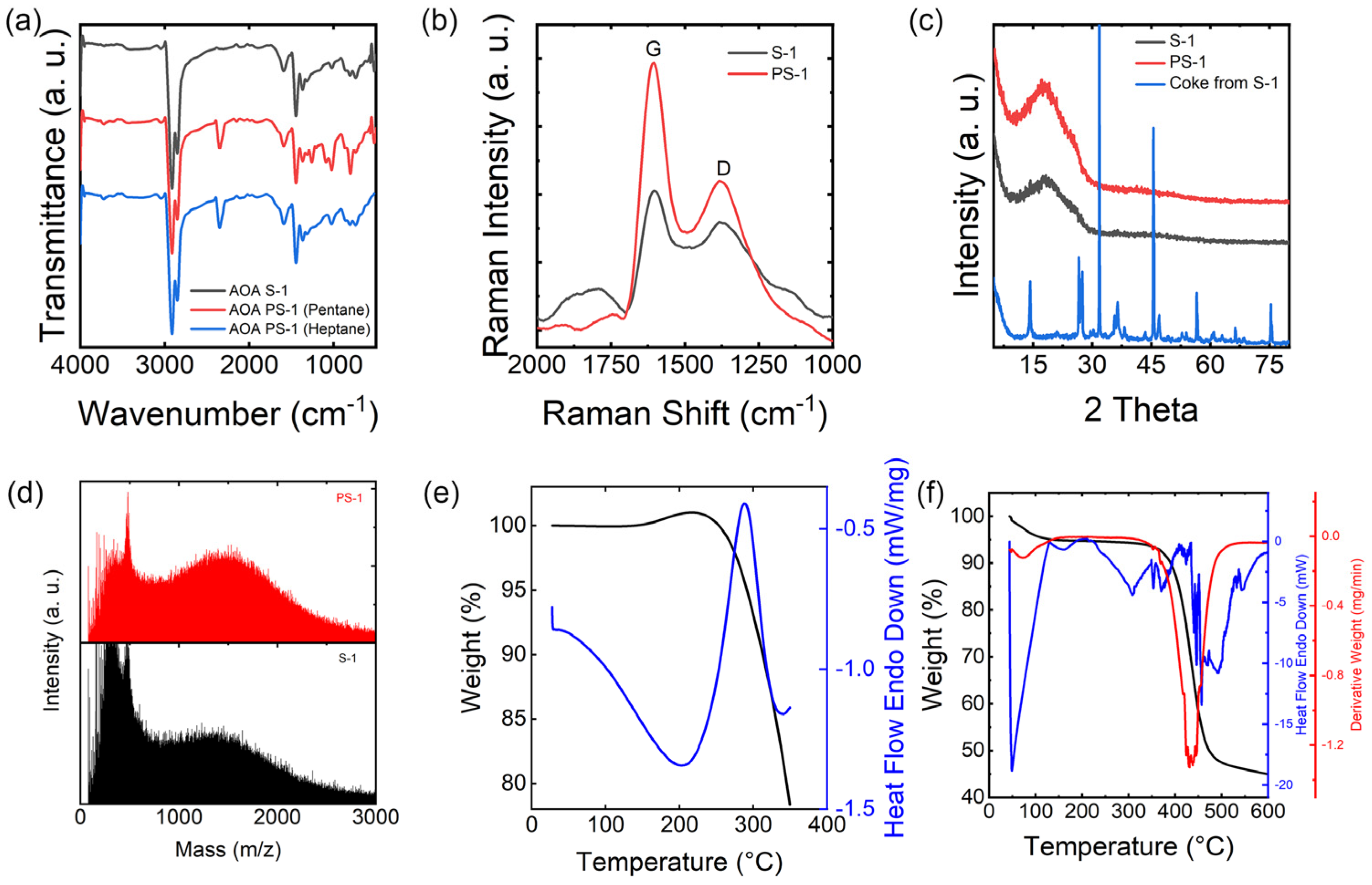
The AOA S−1 feedstock was pretreated and characterized using FT−IR, Raman spectrometry, TGA, and element analysis. The pre−treatment process involved removing volatile light hydrogen carbon and coke from the AOA feedstock. Pentane (C5) was utilized for pretreating the asphaltene in order to remove light hydrogen carbon present in AOA, which may cause defects in the final products, and toluene was used for removing the coke (toluene insoluble fraction), which may block the nozzle during the spinning process.
FT−IR was utilized to examine the functional groups within the molecular structure of asphaltene, which primarily appears in the frequency range of 400–4000 cm
−1. FT−IR spectra of the AOA S−1 before and after solvent pretreatment are shown in
Figure 3a. Feedstock and pretreated AOA are rather similar. All the spectra of pretreated AOA show peaks around 3050 and 1600 cm
−1 which correspond to aromatic stretching of the C−H bond and ring, respectively, and the broad peaks from 920 to 850 cm
−1 correspond to an out−of−plane aromatic structure. Various peaks from 3000 to 2800 cm
−1 are assigned to asymmetric and symmetric stretching, and the peak at 1460 cm
−1 is assigned to asymmetric bending of the aliphatic structure. The bands at 3450 cm
−1 correspond to the stretching vibration of O−H. Heptane (C7) was also used for pretreating the raw materials. The heptane pretreated asphaltene shows a similar FT−IR pattern to that treated by pentane, indicating that the similar volatile (low molecule fraction) was removed. Similar results can be found in element analysis as shown in the
Table 2. For convenience, the pentane−pretreated samples were used for the whole experiment.
Typically, Raman spectrometry is used for carbonaceous materials like asphaltene; the
G band at 1605 cm
−1 and the
D band at 1380 cm
−1 are of particular interest. The
G band corresponds to the stretching vibrations of
sp2 carbon atoms found within both the aromatic hexagonal sheet and the carbon chain. Conversely, the
D band reflects the presence of disordered or less structured
sp3 carbon atoms. In
Figure 3b, AOA S−1 and PS−1 were characterized by Raman spectrometry. It is noted that the
G peaks in both S−1 and PS−1 exhibit sharper features compared to the broader
D peaks. This sharpness suggests a higher degree of short−range order within the aromatic structure of asphaltene. The
ID/
IG ratio serves as an indicator of the proportion of
sp2 carbon atoms in the samples, which play a pivotal role in the stabilization and carbonization processes and are also key to the final mechanical properties. Notably, the
ID/
IG ratios for S−1 and PS−1 are 0.78 and 0.56, respectively. These values suggest a difference in the content of
sp2 carbon atoms, providing insight into the nature of the stabilization and carbonization processes.
XRD is a valuable technique for providing in−depth insights into the level of crystallinity in asphaltene. In conjunction with Raman spectra, this method has been employed to gauge the interlayer spacing within petroleum asphaltene.
Figure 3c displays the XRD patterns of AOA, both before and after pre−treatment. Following pre−treatment, the PS−1 pattern closely resembles that of S−1. Both S−1 and PS−1 exhibit broad peaks at approximately 20°, indicative of the
γ−band and signifying the spacing between alkyl chains or saturated rings. The XRD pattern of the coke produced during the pre−treatment of S−1 displays multiple sharp peaks, which points to the presence of contaminating salts and metals in the raw materials, which were eliminated during the solvent pretreatment process.
As depicted in
Figure 3d, it is evident that the average molecular weight of S−1 is marginally greater than that of PS−1, which corroborates the findings from the FT−IR and CHNS analyses. This underscores the effectiveness of Soxhlet extraction with C5 or C7 in eliminating low molecular weight compounds. Nevertheless, the difference in molecular weight is not particularly substantial, owing to the asphaltene’s inherent tendency to form robust clusters. The molecular weight of both PS−1 and S−1 falls within the range of 500 to 1000 amu, indicating the presence of monomeric asphaltenes with 1–3 ring aromatic chromophores. Meanwhile, the removal of the low molecular weight fraction, often referred to as the volatile fraction, had a notable impact on enhancing the spinning, stabilization, and carbonization processes. This elimination of volatile compounds addressed potential issues related to gas release during spinning and reduced defects during the stabilization and carbonization phases.
The oxygen stabilization process is the most time and energy−intensive step in carbon fiber production. The temperature at which oxygen is absorbed plays a crucial role in achieving a higher degree of stabilization, a fact that can be confirmed through TGA conducted in an ambient air environment. In
Figure 3e, the TGA profiles illustrate the oxygen absorption of AOA during the oxidation stabilization process. The heating rate is set at 2 °C/min, and the experiments are conducted under airflow conditions. For PS−1, there is a continuous decrease in weight as the temperature increases, with distinct stages or increments observed at 220 °C. The stabilization process for PS−1 involves typical reactions such as oxidation, dehydration, condensation, oxidation cross−linking, the elimination of volatile components, and oxidative decomposition. In most cases, the weight of the fibers tends to increase during the stabilization process due to oxygen uptake. The weight of PS−1 started to decrease at around 300 °C, indicating that the decomposition temperature of PS−1 is around 300 °C in air conditions.
A TGA/DSC analysis was carried out in a nitrogen environment to assess the melt spinnability of an asphaltene sample and to determine the appropriate melt−spinning conditions. By examining the TGA/DSC data represented in
Figure 3f for the neat PS−1, it is possible to estimate the operational temperatures for melt spinning. These temperatures fall within the range between the melting point and the temperature at which the sample undergoes degradation. An endotherm peak can be found starting from 220 °C without obvious weight loss, which indicates the melting of PS−1, with degradation commencing around 350 °C. This affords a suitable temperature range for conducting further rheological tests and the melt−spinning process, as it allows operations to occur between the melting range and the degradation temperature without causing sample degradation during the spinning and stabilization processes. It is important to highlight that there exists a temperature differential of approximately 50 °C between the decomposition temperature of PS−1 in air and nitrogen atmospheres. This observation implies that the upper limit for the processing temperature of PS−1 during the spinning process should not exceed 300 °C. Simultaneously, it is imperative to underscore that a preliminary stabilization step, conducted at temperatures below 300 °C, is a prerequisite for the stabilization process.
The elemental analysis results can be found in
Table 2. To determine the composition, solid samples were subjected to combustion at 1800 °C, and the resulting carbon, hydrogen, nitrogen, and sulfur oxides were identified and quantified as weight percentages within the sample. Notably, nitrogen and sulfur are predominantly found within the asphaltene structure rather than in the maltenes. Conversely, the decrease in oxygen content is ascribed to the removal of maltenes. The carbon−to−hydrogen ratio for S−1, PS−1 using pentane, and PS−1 using heptane are 9.91, 10.21, and 9.83, respectively. This observation indicates that PS−1 possesses a higher level of aromaticity compared to S−1, which is necessary for better post−treatment of PS−1 fibers.
Rheology is used to understand the properties of asphaltene melts and to predict the properties of the melt−spinning fiber. Because of the brittleness of the asphaltene, different polymers are considered to serve as additives for modification of the processability of asphaltene. The viscosity of pure PS−1 at various temperatures was determined and is depicted in
Figure 4a. The viscosity of the undiluted PS−1 sample remains approximately constant at around 103 Pa·s across all of the temperatures analyzed under typical shear rates for operation. However, when the shear rate is raised to approximately 10 s
−1, a substantial ten−fold reduction in viscosity is observed. In addition, the rheological characteristics of mixtures comprising both PS (
Figure 4b,c) and SBS (
Figure 4d,e) at concentrations of 5% and 10% were determined with PS−1. This indicates that the viscosity of all mixed samples falls within a similar range (approximately 103 Pa·s) to that of the pure PS−1 sample, at the specified shear rate (approximately 10 s
−1). It is noteworthy that the PS−based blend compositions exhibit a distinct shear−thinning behavior at higher shear rates compared to the SBS−based blends. This observation highlights the potential to operate at lower viscosities while working at higher shear rates. The first normal stress difference of the blended specimens was compared with the pristine, pre−treated sample in
Figure 4f. The first normal stress difference serves as an indicator of the elastic properties within the samples. It is evident that the amalgamation of PS−1 with polymers (PS and SBS) resulted in elevation of the first normal stress difference, signifying an augmented elastic component within the samples. This heightened elasticity in the blended specimens has the potential to enhance the stiffness of the spun fibers.
3.2. Preparation of AOA−CFs through the Use of the Conventional Method
A comprehensive analysis of the morphological characteristics of spun fibers, stabilized fibers, and carbonized fibers, following various thermal post−treatments, was carried out in this study. Spun fibers were obtained using a godet system (
Figure 5a). To avert oxidation, these spun fibers, known for their inherent brittleness, were carefully preserved in a vacuum chamber. The SEM image of spun fibers is shown in
Figure 5b, most of the spun fibers exhibited a diameter of approximately 30 μm, with lengths reaching up to around 1 m. Notably, the spun fibers retained a “groove” structure, a remnant of imperfections stemming from the nozzle during the spinning process. The stabilization of spun PS−1 fibers, a critical step in the production of carbon fibers, typically involves subjecting them to temperatures ranging from 200 to 500 °C in an air or oxygen atmosphere. The TGA in
Figure 3e reveals a broad peak at approximately 220 °C, signifying the onset of oxygen absorption by PS−1. Beyond 300 °C, a sharp reduction in weight is observed, indicating the decomposition of PS−1 in an air environment. For the stabilization of spun PS−1 fibers, conventional methods were employed as part of the control experiment. In the conventional process, the spun PS−1 fibers underwent heat treatment at 220 °C for 4 h, followed by 280 °C for 4 h, with a heating rate of 1 °C/min, while being exposed to a 500 mL/min airflow.
Figure 5c showcases the resulting morphology of the PS−1 fibers after undergoing conventional stabilization. In contrast to the spun fibers, the surface of the stabilized fibers exhibited a smoother and denser texture, attributed to oxygen substitution and cyclization reactions that transpire during the stabilization process [
34]. It is noteworthy that the successful stabilization of PS−1 fibers necessitates the application of tension, as failure to do so may result in their melting and fusion. Following the stabilization process, the stabilized fibers were subsequently subjected to conventional carbonization using a tube furnace. This carbonization process was executed in two steps: firstly at 600 °C for 30 min and then at 1100 °C for an additional 30 min by employing a heating rate of 5 °C/min in an argon gas atmosphere, which was adopted as the control method.
Figure 5d presents the SEM image of the carbonized fibers produced using the conventional approach. Notably, these carbonized fibers exhibited a smoother surface, attributed to the formation of cross−linking carbon–carbon bonds.
Figure 6a displays the XRD patterns for both stabilized PS−1 and carbonized PS−1 fibers. Following the carbonization process, a noticeable enhancement in the intensity of peaks in the XRD pattern of the carbonized fibers was obtained. The XRD pattern of the carbonized fibers revealed the presence of two distinct broad peaks at 26° and 43°, corresponding to the (002) and (100) crystallographic planes, respectively. The (002) plane signifies an increase in the spacing between the aromatic layers, indicative of a higher degree of crystallinity when compared to the stabilized fibers. On the other hand, the (100) plane suggests the presence of graphite layers within the fibers [
35].
Figure 6b presents the Raman spectra for PS−1, stabilized fibers, and carbonized fibers, with specific attention given to the
G band at 1605 cm
−1 and the
D band at 1380 cm
−1. The
G band is associated with the stretching vibrations of
sp2 carbon atoms found within the aromatic hexagonal sheet and the carbon chain, while the
D band signifies the presence of disordered or less structured
sp3 carbon atoms. It is noteworthy that the
G peaks in PS−1, stabilized fibers, and carbonized fibers exhibit sharper features compared to the broader
D peaks. This sharpness indicates a higher degree of short−range order within the asphaltene’s aromatic structure. The
ID/
IG ratio, serving as an indicator of the proportion of
sp2 carbon atoms in the samples, plays a crucial role in understanding the processes of stabilization and carbonization. Notably, the
ID/
IG ratio for PS−1 is 0.56, suggesting variations in the content of
sp2 carbon atoms and offering insights into the nature of the stabilization and carbonization procedures. The
ID/
IG ratio for stabilized fibers is 0.63, while it increases to 0.69 for the carbonized PS−1 fibers. This rise in the ratio indicates an augmentation in the proportion of
sp2 carbon atoms after stabilization and subsequent carbonization, consistent with the findings from the XRD and elemental analyses. These results emphasize the importance of strict temperature to enhance the
sp2 ratio in carbon fibers.
The crystallite parameters of graphite crystals of carbon fibers were calculated by using Bragg’s law and the Debye–Scherrer equation, as presented in
Table 3 [
36]. The interlayer spacing (
d(002)), the apparent crystallite thickness (
Lc), and the apparent layer−plane length parallel to the fiber axis (
La) are calculated based on the following equations from
β:
where
d(002) is the interlayer spacing,
θ is the Bragg angle of the peaks,
λ is the wavelength of the X−ray used (0.154 nm), and
β is the full width at half maximum (FWHM). The form factor
K is 0.89 for
Lc and 1.84 for
La, respectively. As presented in
Table 3, the interlayer spacing (
d(002)) of AOA carbon fibers indicates a similar ordered structure compared with conventional carbon fibers.
Furthermore, as depicted in
Table 4, the carbon content exhibited an increase from 74.10% to 87.58% for stabilization and carbonized fibers, respectively, while the H/C ratio decreased from 0.081103 to 0.003540. This serves as further evidence of the formation of carbon basal planes during the carbonization process [
37]. The reduction in the levels of H, N, and S suggests the elimination of heteroatoms during carbonization. Notably, even after carbonization, the sulfur content still stands at 5.56%, potentially introducing defects into the fibers. The method for effectively removing sulfur from the conventional carbonization process remains unclear.
3.3. Preparation of AOA−CFs by Microwave Plasma
Figure 7 presents the SEM images depicting a comparison between conventionally stabilized fibers before and after microwave plasma treatment. After subjecting the fibers to 5 min microwave plasma treatment at 1000 W, evident surface damage and the presence of pores were observed, as illustrated in
Figure 7b. This surface degradation can be attributed to the physical and chemical interactions, as well as collisions, between the charged particles within the plasma and the surface of the stabilized fibers [
23]. The observed defects resulting from the microwave plasma treatment indicate the necessity for more robust and stable fibers when undergoing such treatment.
For the assessment of microwave plasma treatment, carbonized fibers, characterized by their enhanced robustness and stability, were selected as the subject of investigation.
Figure 8 presents the morphological analysis of conventionally carbonized fibers following treatment with microwave plasma at varying time intervals. Comparing the level of damage observed in the microwave plasma treatment between stabilized fibers (
Figure 7b) and conventionally carbonized fibers, it is noted that the damage to the carbonized fibers was relatively less pronounced. As the treatment time extended from 5 to 7 and then to 9 min, a noticeable increase in surface imperfections, such as dents, was observed, as depicted in
Figure 8a–c. This escalation in the number of defects may be attributed to the ongoing attack by argon ions on the fiber [
38]. In particular, fibers treated with microwave plasma for 9 min (
Figure 8c) exhibited more substantial surface damage, which could potentially impact the mechanical properties of the final carbon fiber products.
Figure 9 illustrates the XRD pattern and Raman spectra of carbonized fibers subjected to microwave plasma treatment for durations of 5, 7, and 9 min. In
Figure 9a, it is evident that the peak around 26°, corresponding to the (002) plane, exhibited increased sharpness following microwave plasma treatments. This sharpening of the peak indicates an augmentation in the spacing between the aromatic layers, indicative of a higher degree of crystallinity compared to conventionally carbonized fibers. Key crystalline parameters for conventional carbonized fibers before and after microwave plasma treatment are detailed in
Table 3. The interlayer spacing (
d) decreased to 0.360 nm, while the crystallite parameters of graphite thickness
Lc and
La increased with longer plasma treatment durations. These findings suggest that microwave plasma treatment fosters the transition from a turbostratic graphite−like structure to a more ordered graphite structure [
39]. In
Figure 9b, the Raman spectra of carbonized fibers subjected to microwave plasma treatment for 5, 7, and 9 min are presented. With increasing treatment duration, there was a noticeable change in the intensity of both the
G and
D peaks, with the
G peak becoming relatively more pronounced. The
ID/
IG ratios of 5, 7, and 9 min treatment are 0.73, 0.68, and 0.64, respectively, as the treatment time increased, signifying a higher proportion of ordered graphitic carbon with
sp2 hybridization [
34]. Notably, despite an extended treatment duration of up to 9 min, as displayed in
Figure 9b, the presence of the
D peak suggests the persistence of defects and disordered graphite structures within the carbon fiber [
39]. More strict conditions, such as a higher temperature, are needed to increase the
ID/
IG ratio to further improve the mechanical performance of the final product.
Table 5 presents the element analysis before and after microwave plasma treatment. A comparison with conventionally carbonized fibers reveals that prolonged microwave plasma treatment, especially over a 9 min duration, led to an increase in the carbon content from 87.58% to 91.84%. This heightened carbon content signifies a reduction in heteroatoms and an improvement in the carbon–carbon structure, directly correlating to enhanced mechanical performance. The content of all heteroatoms, including hydrogen, nitrogen, sulfur, and oxygen, experienced a decrease subsequent to microwave plasma treatment. This reduction indicates the effectiveness of microwave plasma in eliminating heteroatoms. Particularly challenging is the reduction in sulfur content, given its inheritance from the original oilsands. Microwave plasma demonstrates promise in achieving higher temperatures in shorter durations and in effectively eliminating heteroatoms during the fabrication of asphaltene−based carbon fibers.
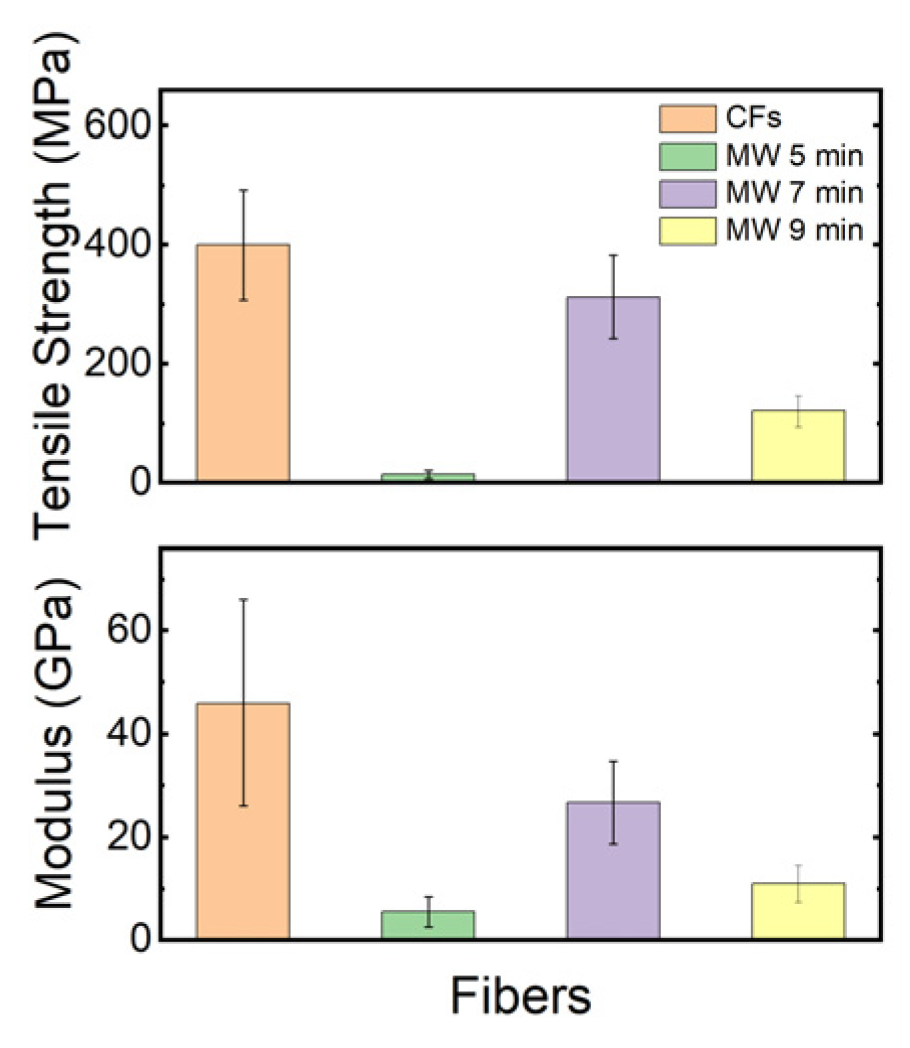
The tensile strength and modulus of conventionally carbonized fibers and microwave plasma−treated fibers were measured and are shown in
Figure 10. The results indicate that after carbonization using microwave plasma, the average tensile strength and modulus values have been reduced, which can be backed up by the surface damage on the fibers, as seen in
Figure 7, and the rapid heating rate induced by the microwave plasma. The presence of voids in the fiber morphology is undesirable as it renders these fibers unsuitable for structural applications [
38]. These values are lower than those reported in the previous literature, suggesting potential areas for process optimization such as melt−spinning, stabilization, or carbonization to improve mechanical properties further.
3.4. Modelling of MW Plasma
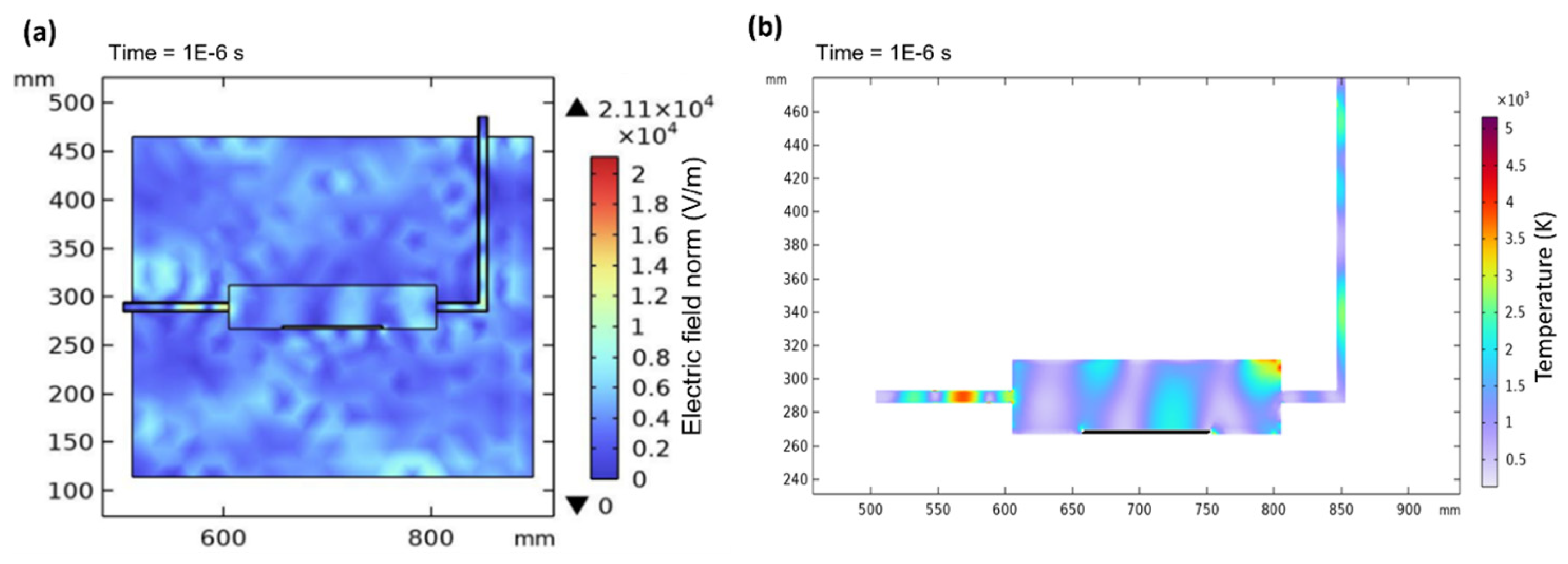
Figure 11a shows the electric field norm in the microwave cavity, while
Figure 11b displays the contour temperature plot of the plasma temperature in the quartz tube at 1E−6 s. When a microwave power of 1000 W is applied, there is instability in the electric field distribution due to interference with surroundings in the cavity. However, once the electric field stabilizes and becomes continuous at 10
−6 s, electromagnetic waves propagate and excite argon gas to generate plasma. This leads to stable and consistent plasma generation inside the quartz tube which immediately elevates the temperature, as shown in
Figure 11b. The inconsistency in temperature within the quartz tube can be attributed to factors such as random particle collisions, variations in plasma density, and interactions between wave modes and argon gas [
38].
Successful extraction of the high molecular weight fraction from Alberta oilsands was achieved through solvent−based techniques, resulting in the successful fabrication of carbon fibers. Notably, the asphaltene component exhibited brittle characteristics and high viscosity during the spinning process, posing significant challenges for processing. However, the addition of polymer additives effectively altered the viscosity of the raw asphaltene, enhancing its processability. These polymer additives interacted with asphaltene molecules, leading to modification of viscosity and enabling precise control of melt spinnability. The subsequent post−treatment of asphaltene fibers bore similarities to the procedures applied to pitch carbon fibers. The stabilization process involved oxygen absorption and the conversion of gaseous emissions, transforming the fibers from a thermoplastic state to a thermoset state. During the carbonization process, the removal of heteroatoms occurred, with higher temperatures contributing to greater carbon content and a reduced presence of heteroatoms. Nonetheless, certain heteroatoms, including sulfur and nitrogen, remained within the final carbonized fibers, firmly embedded in the molecular bonds and originating from the oilsands source. This marked a distinction from mesophase pitch carbon fibers, which are derived from naphthalene. Furthermore, the potential of microwave plasma technology showed promise in further reducing heteroatoms within asphaltene carbon fibers, although further investigation and confirmation are necessary for future research endeavors.
The processing of oilsands asphaltene remains challenging due to variations in source and location. These differences result in distinct physical and chemical properties, particularly in terms of viscosity, which hinders consistent manufacturing. Moreover, the presence of heteroatoms such as sulfur and nitrogen in asphaltene proves difficult to eliminate, thus impeding the production of asphaltene carbon fibers and affecting the mechanical properties of the final carbon fiber product. The brittleness of asphaltene further complicates continuous fiber manufacturing. The quest for cost−effective and efficient polymer additives to enhance the melt spinnability of asphaltene is an ongoing investigation. Given the intricate structure of asphaltene, the mechanism for modifying its properties is still the subject of ongoing research. Comparatively, the mechanical properties of the final asphaltene carbon fibers fall short when measured against commercial carbon fibers derived from PAN and mesophase pitch. Addressing this disparity remains a pertinent challenge in this field.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}