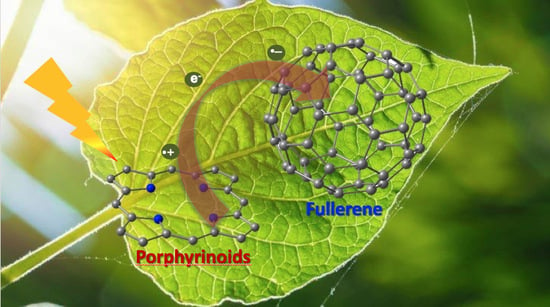
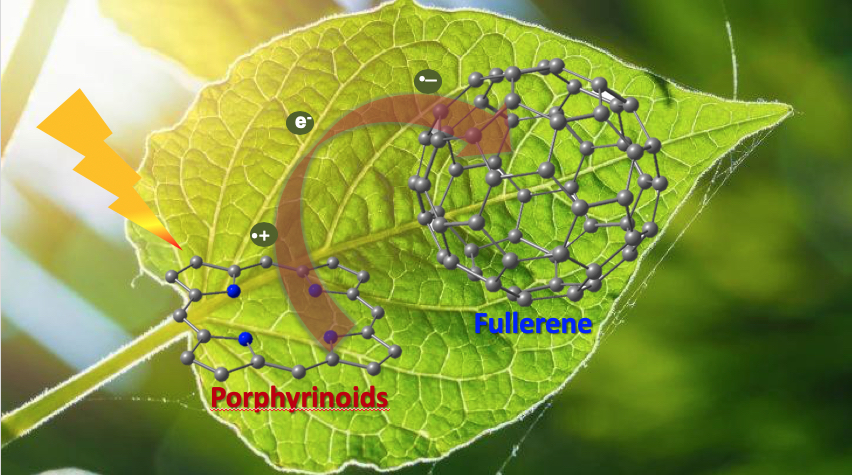
Porphyrinoid–Fullerene Hybrids as Candidates in Artificial Photosynthetic Schemes

 , , and
, , and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
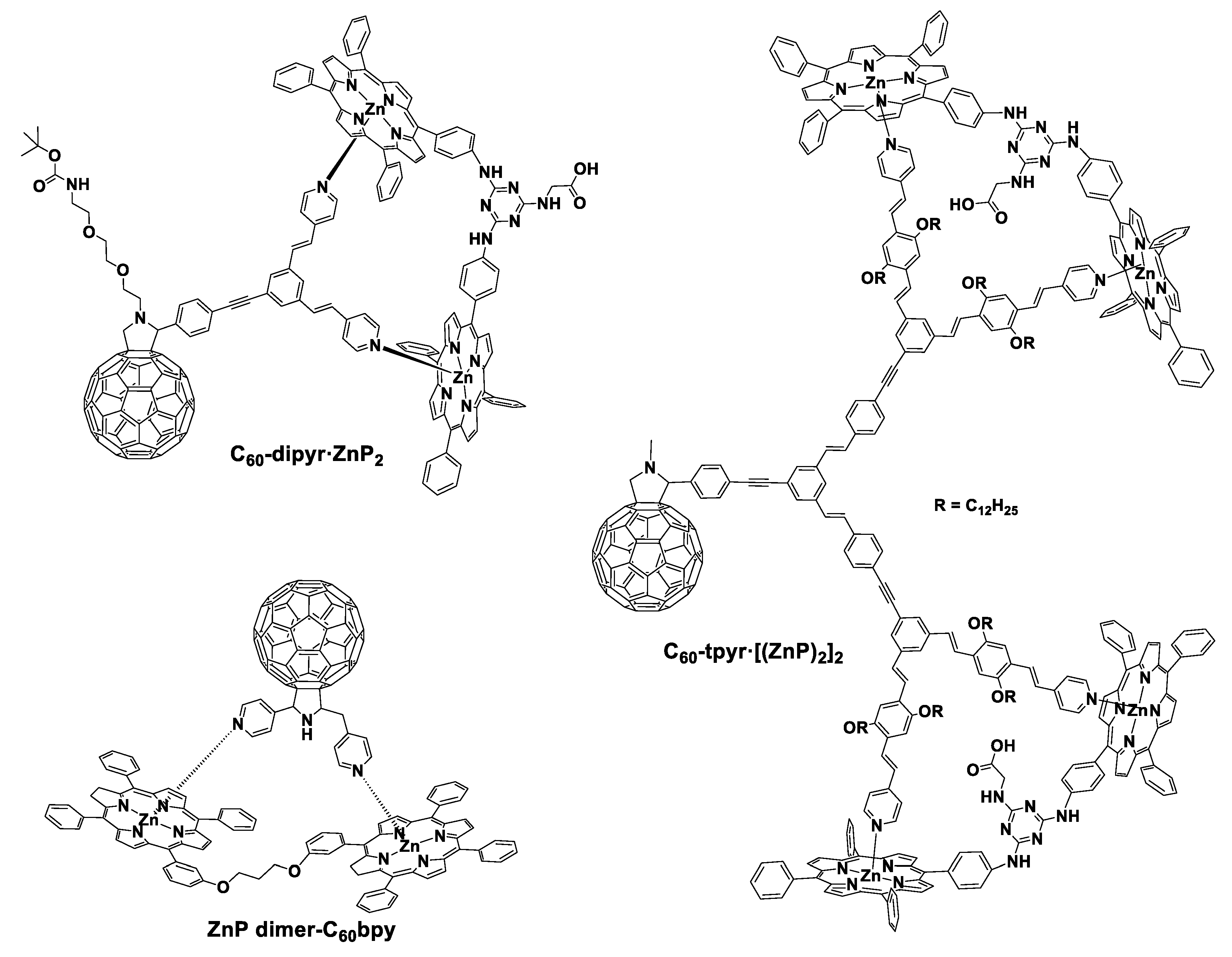
2. Porphyrinoids Connected with Fullerenes
2.1. Covalent and Noncovalent Connected Porphyrinoid–Fullerene Assemblies
2.2. The Influence of the Distance Between Porphyrinoids and C60
2.3. Increasing the Number of Porphyrin Derivatives in Porphyrin–C60 Entities
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Tachibana, Y.; Vayssieres, L.; Durrant, J.R. Artificial photosynthesis for solar water-splitting. Nat. Photonics 2012, 6, 511. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, L. Artificial photosynthesis: opportunities and challenges of molecular catalysts. Chem. Soc. Rev. 2019, 48, 2216–2264. [Google Scholar] [CrossRef] [PubMed]
- Bredas, J.L.; Sargent, E.H.; Scholes, G.D. Photovoltaic concepts inspired by coherence effects in photosynthetic systems. Nature Mater. 2017, 16, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-W.; Zhong, D.-C.; Lu, T.-B. Artificial photosynthesis: Catalytic water oxidation and CO2 reduction by dinuclear non-noble-metal molecular catalysts. Coord. Chem. Rev. 2018, 377, 225–236. [Google Scholar] [CrossRef]
- Fukuzumi, S. Development of bioinspired artificial photosynthetic systems. Phys. Chem. Chem. Phys. 2008, 10, 2283–2297. [Google Scholar] [CrossRef]
- Rudolf, M.; Kirner, S.V.; Guldi, D.M. A multicomponent molecular approach to artificial photosynthesis—The role of fullerenes and endohedral metallofullerenes. Chem. Soc. Rev. 2016, 45, 612–630. [Google Scholar] [CrossRef]
- Bottari, G.; Suanzes, J.A.; Trukhina, O.; Torres, T. Phthalocyanine−carbon nanostructure materials assembled through supramolecular interactions. J. Phys. Chem. Lett. 2011, 2, 905–913. [Google Scholar] [CrossRef]
- Hiroto, S.; Miyake, Y.; Shinokubo, H. Synthesis and Functionalization of Porphyrins through Organometallic Methodologies. Chem. Rev. 2017, 117, 2910–3043. [Google Scholar] [CrossRef]
- Chen, Y.; Royal, G.; Flahaut, E.; Cobo, S.; Bouchiat, V.; Marty, L.; Bendiab, N. Light control of charge transfer and excitonic transitions in a carbon nanotube/porphyrin hybrid. Adv. Mater. 2017, 29, 1605745. [Google Scholar] [CrossRef]
- Zhang, W.; Lai, W.; Cao, R. Energy-related small molecule activation reactions: oxygen reduction and hydrogen and oxygen evolution reactions catalyzed by porphyrin- and corrole-based systems. Chem. Rev. 2017, 117, 3717–3797. [Google Scholar] [CrossRef]
- Ladomenou, K.; Natali, M.; Iengo, E.; Charalampidis, G.; Scandola, F.; Coutsolelos, A.G. Photochemical hydrogen generation with porphyrin-based systems. Coord. Chem. Rev. 2015, 304–305, 38–54. [Google Scholar] [CrossRef]
- Pascal, S.; Bucher, L.; Desbois, N.; Bucher, C.; Andraud, C.; Gros, C.P. Synthesis, electrochemistry, and photophysics of Aza-BODIPY porphyrin dyes. Chem. Eur. J. 2016, 22, 4971–4979. [Google Scholar] [CrossRef] [PubMed]
- Hebié, S.; Dimé, A.K.D.; Devillers, C.H.; Lucas, D. Electrochemistry as an attractive and effective tool for the synthesis and immobilization of porphyrins on an electrode surface. Chem. Eur. J. 2015, 21, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Ladomenou, K.; Nikolaou, V.; Charalambidis, G.; Coutsolelos, A.G. Artificial hemes for DSSC and/or BHJ applications. Dalton Trans. 2016, 45, 1111–1126. [Google Scholar] [CrossRef]
- Bottari, G.; Trukhina, O.; Ince, M.; Torres, T. Towards artificial photosynthesis: Supramolecular, donor–acceptor, porphyrin- and phthalocyanine/carbon nanostructure ensembles. Coord. Chem. Rev. 2012, 256, 2453–2477. [Google Scholar] [CrossRef]
- Guldi, D.M. Fullerene-porphyrin architectures; photosynthetic antenna and reaction center models. Chem. Soc. Rev. 2002, 31, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Frischmann, P.D.; Mahata, K.; Würthner, F. Powering the future of molecular artificial photosynthesis with light-harvesting metallosupramolecular dye assemblies. Chem. Soc. Rev. 2013, 42, 1847–1870. [Google Scholar] [CrossRef]
- Collavini, S.; Delgado, J.L. Fullerenes: the stars of photovoltaics. Sustain. Energy Fuels 2018, 2, 2480–2493. [Google Scholar] [CrossRef]
- Imahori, H.; Guldi, D.M.; Tamaki, K.; Yoshida, Y.; Luo, C.; Sakata, Y.; Fukuzumi, S. Charge separation in a novel artificial photosynthetic reaction center lives 380 ms. J. Am. Chem. Soc. 2001, 123, 6617–6628. [Google Scholar] [CrossRef]
- Zieleniewska, A.; Lodermeyer, F.; Roth, A.; Guldi, D.M. Fullerenes—How 25 years of charge transfer chemistry have shaped our understanding of (interfacial) interactions. Chem. Soc. Rev. 2018, 47, 702–714. [Google Scholar] [CrossRef]
- D’Souza, F.; Ito, O. Photoinduced electron transfer in supramolecular systems of fullerenes functionalized with ligands capable of binding to zinc porphyrins and zinc phthalocyanines. Coord. Chem. Rev. 2005, 249, 1410–1422. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Lyubovskaya, R.N. Fullerene complexes with coordination assemblies of metalloporphyrins and metal phthalocyanines. Coord. Chem. Rev. 2014, 262, 16–36. [Google Scholar] [CrossRef]
- Kc, C.B.; D’Souza, F. Design and photochemical study of supramolecular donor–acceptor systems assembled via metal–ligand axial coordination. Coord. Chem. Rev. 2016, 322, 104–141. [Google Scholar] [CrossRef]
- Bottari, G.; de la Torre, G.; Guldi, D.M.; Torres, T. Covalent and noncovalent phthalocyanine-carbon nanostructure systems: Synthesis, photoinduced electron transfer, and application to molecular photovoltaics. Chem. Rev. 2010, 110, 6768–6816. [Google Scholar] [CrossRef] [PubMed]
- Ito, O. Photosensitizing electron transfer processes of fullerenes, carbon nanotubes, and carbon nanohorns. Chem. Rec. 2017, 17, 326–362. [Google Scholar] [CrossRef]
- Nikolaou, V.; Plass, F.; Planchat, A.; Charisiadis, A.; Charalambidis, G.; Angaridis, P.A.; Kahnt, A.; Odobel, F.; Coutsolelos, A.G. Effect of the triazole ring in zinc porphyrin-fullerene dyads on the charge transfer processes in NiO-based devices. Phys. Chem. Chem. Phys. 2018, 20, 24477–24489. [Google Scholar] [CrossRef] [PubMed]
- Huisgen, R. 1,3-Dipolar cycloadditions. Past and future. Angew. Chem. Int. Ed. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Ladomenou, K.; Nikolaou, V.; Charalambidis, G.; Coutsolelos, A.G. "Click"-reaction: An alternative tool for new architectures of porphyrin based derivatives. Coord. Chem. Rev. 2016, 306, 1–42. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- de Miguel, G.; Wielopolski, M.; Schuster, D.I.; Fazio, M.A.; Lee, O.P.; Haley, C.K.; Ortiz, A.L.; Echegoyen, L.; Clark, T.; Guldi, D.M. Triazole bridges as versatile linkers in electron donor-acceptor conjugates. J. Am. Chem. Soc. 2011, 133, 13036–13054. [Google Scholar] [CrossRef]
- Rotas, G.; Charalambidis, G.; Glatzl, L.; Gryko, D.T.; Kahnt, A.; Coutsolelos, A.G.; Tagmatarchis, N. A corrole-azafullerene dyad: Synthesis, characterization, electronic interactions and photoinduced charge separation. Chem. Commun. 2013, 49, 9128–9130. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, F.; Chitta, R.; Ohkubo, K.; Tasior, M.; Subbaiyan, N.K.; Zandler, M.E.; Rogacki, M.K.; Gryko, D.T.; Fukuzumi, S. Corrole-Fullerene Dyads: Formation of Long-Lived Charge-Separated States in Nonpolar Solvents. J. Am. Chem. Soc. 2008, 130, 14263–14272. [Google Scholar] [CrossRef] [PubMed]
- Stangel, C.; Schubert, C.; Kuhri, S.; Rotas, G.; Margraf, J.T.; Regulska, E.; Clark, T.; Torres, T.; Tagmatarchis, N.; Coutsolelos, A.G.; et al. Tuning the reorganization energy of electron transfer in supramolecular ensembles—metalloporphyrin, oligophenylenevinylenes, and fullerene—and the impact on electron transfer kinetics. Nanoscale 2015, 7, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Krug, M.; Stangel, C.; Schubert, C.; Clark, T.; Torres, T.; Coutsolelos, A.G.; Guldi, D.M. Combining zinc phthalocyanines, oligo(p-phenylenevinylenes), and fullerenes to impact reorganization energies and attenuation factors. ChemPhysChem 2019, accepted. [Google Scholar] [CrossRef]
- Guldi, D.M.; Gouloumis, A.; Vazquez, P.; Torres, T.; Georgakilas, V.; Prato, M. Nanoscale organization of a phthalocyanine-fullerene system: Remarkable stabilization of charges in photoactive 1-D nanotubules. J. Am. Chem. Soc. 2005, 127, 5811–5813. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, F.; Deviprasad, G.R.; Zandler, M.E.; Hoang, V.T.; Klykov, A.; VanStipdonk, M.; Perera, A.; El-Khouly, M.E.; Fujitsuka, M.; Ito, O. Spectroscopic, electrochemical, and photochemical studies of self-assembled via axial coordination zinc porphyrin-fulleropyrrolidine dyads. J. Phys. Chem. A 2002, 106, 3243–3252. [Google Scholar] [CrossRef]
- El-Khouly, M.E.; Rogers, L.M.; Zandler, M.E.; Suresh, G.; Fujitsuka, M.; Ito, O.; D’Souza, F. Studies on intra-supramolecular and intermolecular electron-transfer processes between zinc naphthalocyanine and imidazole-appended fullerene. ChemPhysChem 2003, 4, 474–481. [Google Scholar] [CrossRef]
- Follana-Berna, J.; Seetharaman, S.; Martin-Gomis, L.; Charalambidis, G.; Trapali, A.; Karr, P.A.; Coutsolelos, A.G.; Fernandez-Lazaro, F.; D’Souza, F.; Sastre-Santos, A. Supramolecular complex of a fused zinc phthalocyanine-zinc porphyrin dyad assembled by two imidazole-C-60 units: Ultrafast photoevents. Phys. Chem. Chem. Phys. 2018, 20, 7798–7807. [Google Scholar] [CrossRef]
- Seetharaman, S.; Follana-Berna, J.; Martin-Gomis, L.; Charalambidis, G.; Trapali, A.; Karr, P.A.; Coutsolelos, A.G.; Fernandez-Lazaro, F.; Sastre-Santos, A.; D’Souza, F. Sequential, Ultrafast Energy Transfer and Electron Transfer in a Fused Zinc Phthalocyanine-free-base Porphyrin-C-60 Supramolecular Triad. ChemPhysChem 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Segura, J.L.; Giacalone, F.; Gomez, R.; Martin, N.; Guldi, D.M.; Luo, C.P.; Swartz, A.; Riedel, I.; Chirvase, D.; Parisi, J.; et al. Design, synthesis and photovoltaic properties of [60]fullerene based molecular materials. Mater. Sci. Eng. C 2005, 25, 835–842. [Google Scholar] [CrossRef]
- Goransson, E.; Boixel, J.; Fortage, J.; Jacquemin, D.; Becker, H.C.; Blart, E.; Hammarstrom, L.; Odobel, F. Long-Range Electron Transfer in Zinc-Phthalocyanine-Oligo(Phenylene-ethynylene)-Based Donor-Bridge-Acceptor Dyads. Inorg. Chem. 2012, 51, 11500–11512. [Google Scholar] [CrossRef] [PubMed]
- Donhauser, Z.J.; Mantooth, B.A.; Kelly, K.F.; Bumm, L.A.; Monnell, J.D.; Stapleton, J.J.; Price, D.W.; Rawlett, A.M.; Allara, D.L.; Tour, J.M.; et al. Conductance switching in single molecules through conformational changes. Science 2001, 292, 2303–2307. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.B.; Reichert, J.; Weigend, F.; Ochs, R.; Beckmann, D.; Mayor, M.; Ahlrichs, R.; von Lohneysen, H. Electronic transport through single conjugated molecules. Chem. Phys. 2002, 281, 113–125. [Google Scholar] [CrossRef]
- Andréasson, J.; Kodis, G.; Ljungdahl, T.; Moore, A.L.; Moore, T.A.; Gust, D.; Mårtensson, J.; Albinsson, B. Photoinduced Hole Transfer from the Triplet State in a Porphyrin-Based Donor−Bridge−Acceptor System. J. Phys. Chem. A 2003, 107, 8825–8833. [Google Scholar] [CrossRef]
- Yang, S.I.; Lammi, R.K.; Prathapan, S.; Miller, M.A.; Seth, J.; Diers, J.R.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Synthesis and excited-state photodynamics of perylene-porphyrin dyads Part 3. Effects of perylene, linker, and connectivity on ultrafast energy transfer. J. Mater. Chem. 2001, 11, 2420–2430. [Google Scholar] [CrossRef]
- Redmore, N.P.; Rubtsov, I.V.; Therien, M.J. Synthesis, electronic structure, and electron transfer dynamics of (aryl)ethynyl-bridged donor-acceptor systems. J. Am. Chem. Soc. 2003, 125, 8769–8778. [Google Scholar] [CrossRef]
- Stangel, C.; Charisiadis, A.; Zervaki, G.E.; Nikolaou, V.; Charalambidis, G.; Kahnt, A.; Rotas, G.; Tagmatarchis, N.; Coutsolelos, A.G. Case Study for Artificial Photosynthesis: Noncovalent Interactions between C-60-Dipyridyl and Zinc Porphyrin Dimer. J. Phys. Chem. C 2017, 121, 4850–4858. [Google Scholar] [CrossRef]
- Stangel, C.; Plass, F.; Charisiadis, A.; Giannoudis, E.; Chararalambidis, G.; Karikis, K.; Rotas, G.; Zervaki, G.E.; Lathiotakis, N.N.; Tagmatarchis, N.; et al. Interfacing tetrapyridyl-C-60 with porphyrin dimers via -conjugated bridges: artificial photosynthetic systems with ultrafast charge separation. Phys. Chem. Chem. Phys. 2018, 20, 21269–21279. [Google Scholar] [CrossRef]
- D’Souza, F.; Gadde, S.; Zandler, M.E.; Itou, M.; Araki, Y.; Ito, O. Supramolecular complex composed of a covalently linked zinc porphyrin dimer and fulleropyrrolidine bearing two axially coordinating pyridine entities. Chem. Commun. 2004. [Google Scholar] [CrossRef]
- Kc, C.B.; Ohkubo, K.; Karr, P.A.; Fukuzumi, S.; D’Souza, F. A ‘two-point’ bound zinc porphyrin-zinc phthalocyanine-fullerene supramolecular triad for sequential energy and electron transfer. Chem. Commun. 2013, 49, 7614–7616. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolaou, V.; Charisiadis, A.; Stangel, C.; Charalambidis, G.; Coutsolelos, A.G. Porphyrinoid–Fullerene Hybrids as Candidates in Artificial Photosynthetic Schemes. C 2019, 5, 57. https://doi.org/10.3390/c5030057
Nikolaou V, Charisiadis A, Stangel C, Charalambidis G, Coutsolelos AG. Porphyrinoid–Fullerene Hybrids as Candidates in Artificial Photosynthetic Schemes. C. 2019; 5(3):57. https://doi.org/10.3390/c5030057
Chicago/Turabian StyleNikolaou, Vasilis, Asterios Charisiadis, Christina Stangel, Georgios Charalambidis, and Athanassios G. Coutsolelos. 2019. "Porphyrinoid–Fullerene Hybrids as Candidates in Artificial Photosynthetic Schemes" C 5, no. 3: 57. https://doi.org/10.3390/c5030057
APA StyleNikolaou, V., Charisiadis, A., Stangel, C., Charalambidis, G., & Coutsolelos, A. G. (2019). Porphyrinoid–Fullerene Hybrids as Candidates in Artificial Photosynthetic Schemes. C, 5(3), 57. https://doi.org/10.3390/c5030057