
Genome-Wide Identification and Characterization of Sucrose Metabolism Genes Involved in Actinidia rufa and Their Expression Profiling during the Fruit Developmental Stages

Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Sucrose Metabolism Genes in the A. rufa Genome
2.2. Phylogenetic Analysis
2.3. Gene Structure Construction, Conserved Motifs, and Cis-Acting Elements Identification
2.4. Subcellular Localization and Protein Physicochemical Properties Analysis
2.5. Collinearity and Selective Pressure Analysis
2.6. Transcriptome Profile Analysis
3. Results
3.1. Genome-Wide Identification of Sucrose Metabolism-Related Gene Families from A. rufa
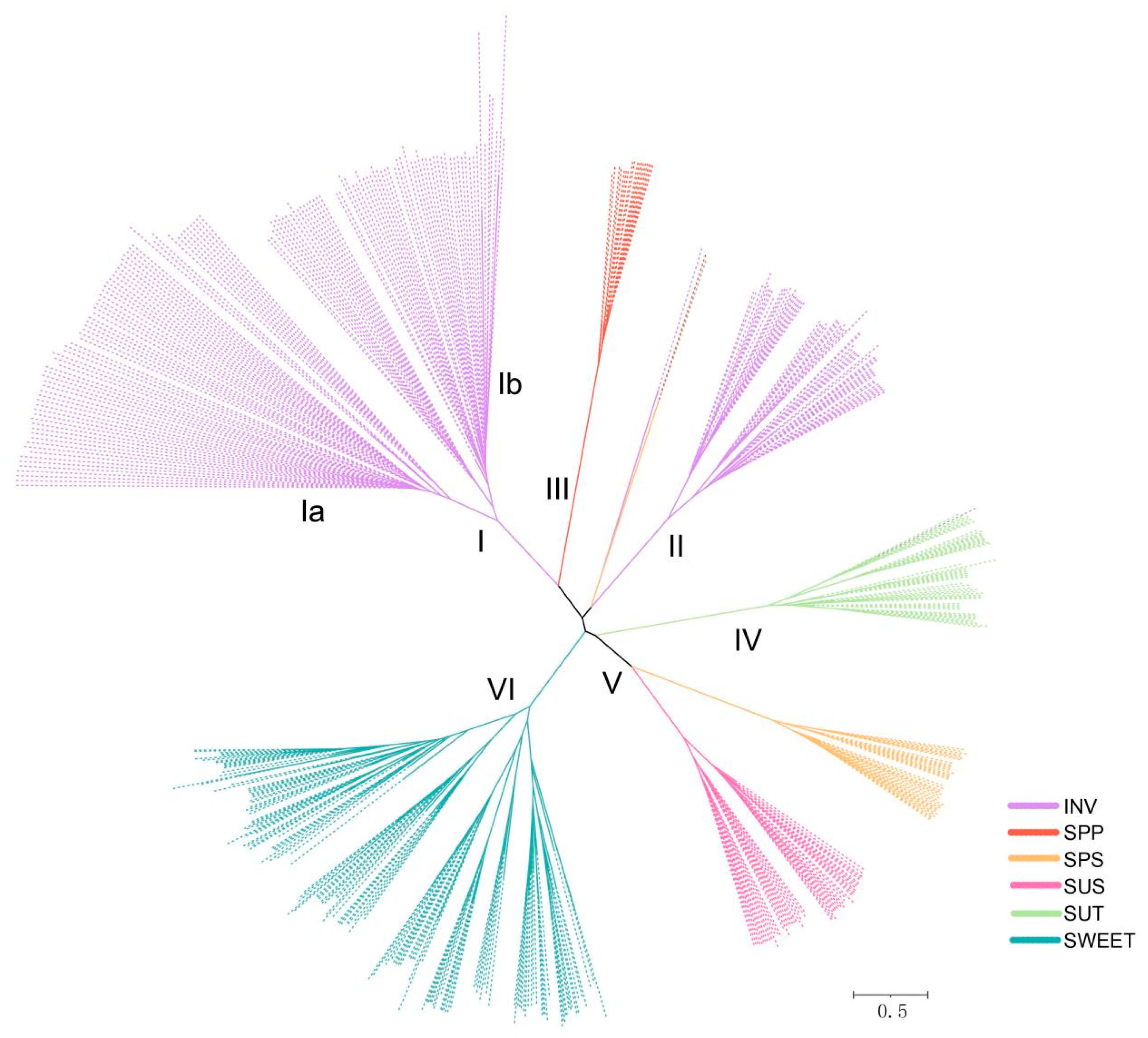
3.2. Phylogenetic Analysis
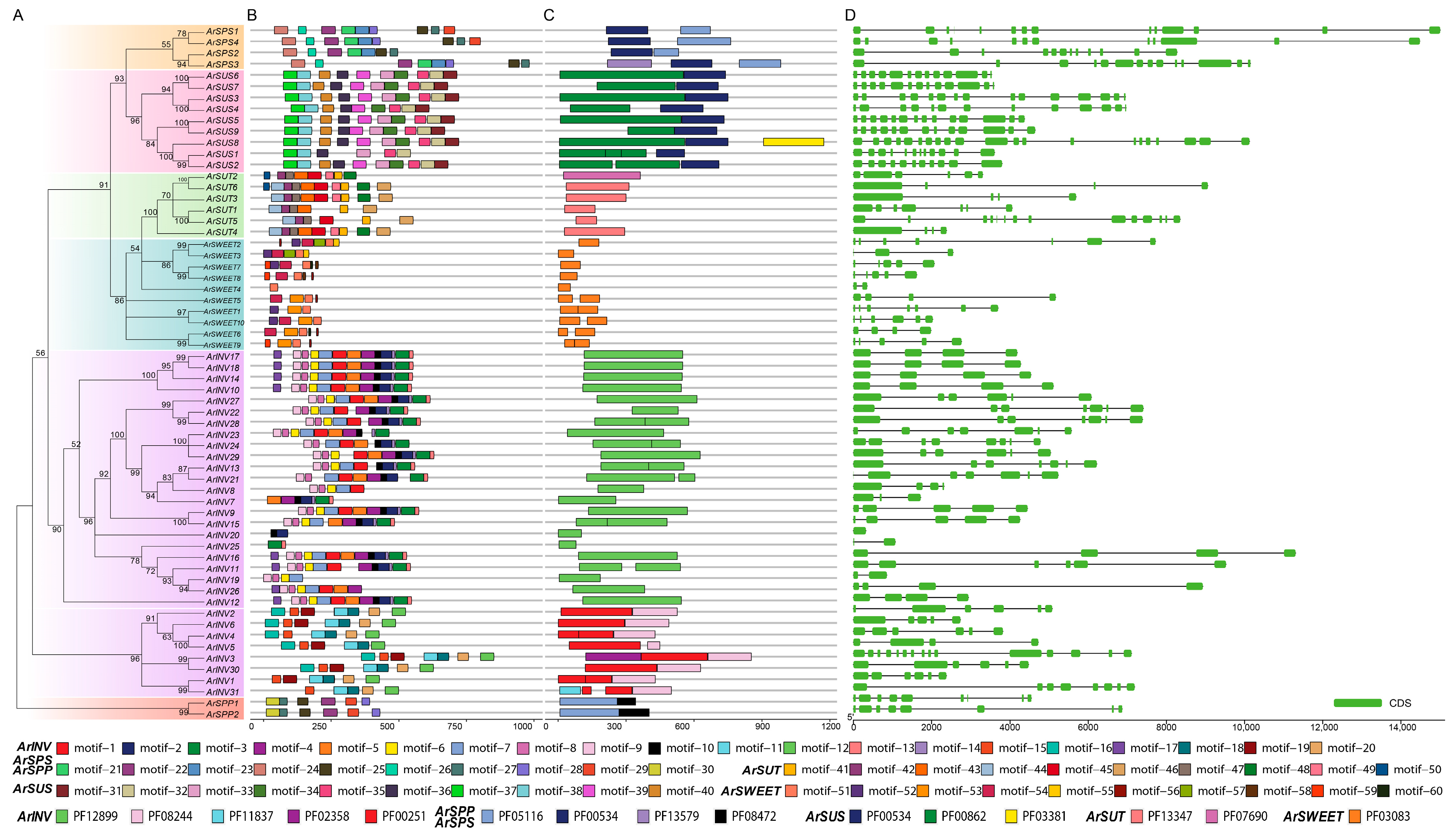
3.3. Gene Structure, Conserved Motif, and Protein Domain Analysis
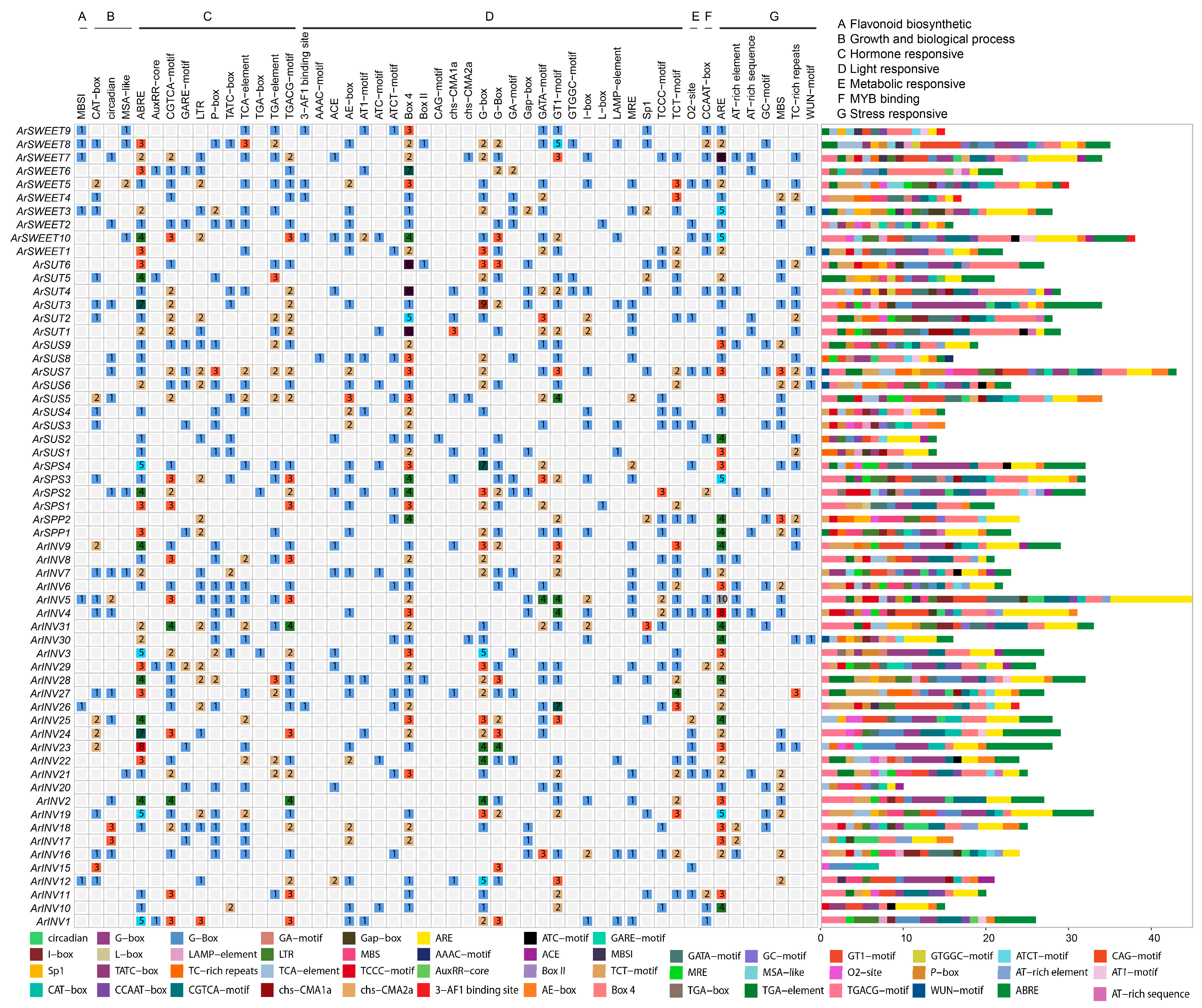
3.4. Promoter Cis-Elements Analysis of Sucrose Metabolism Genes
3.5. Collinearity and Selective Pressure Analysis of Sucrose Metabolism Genes
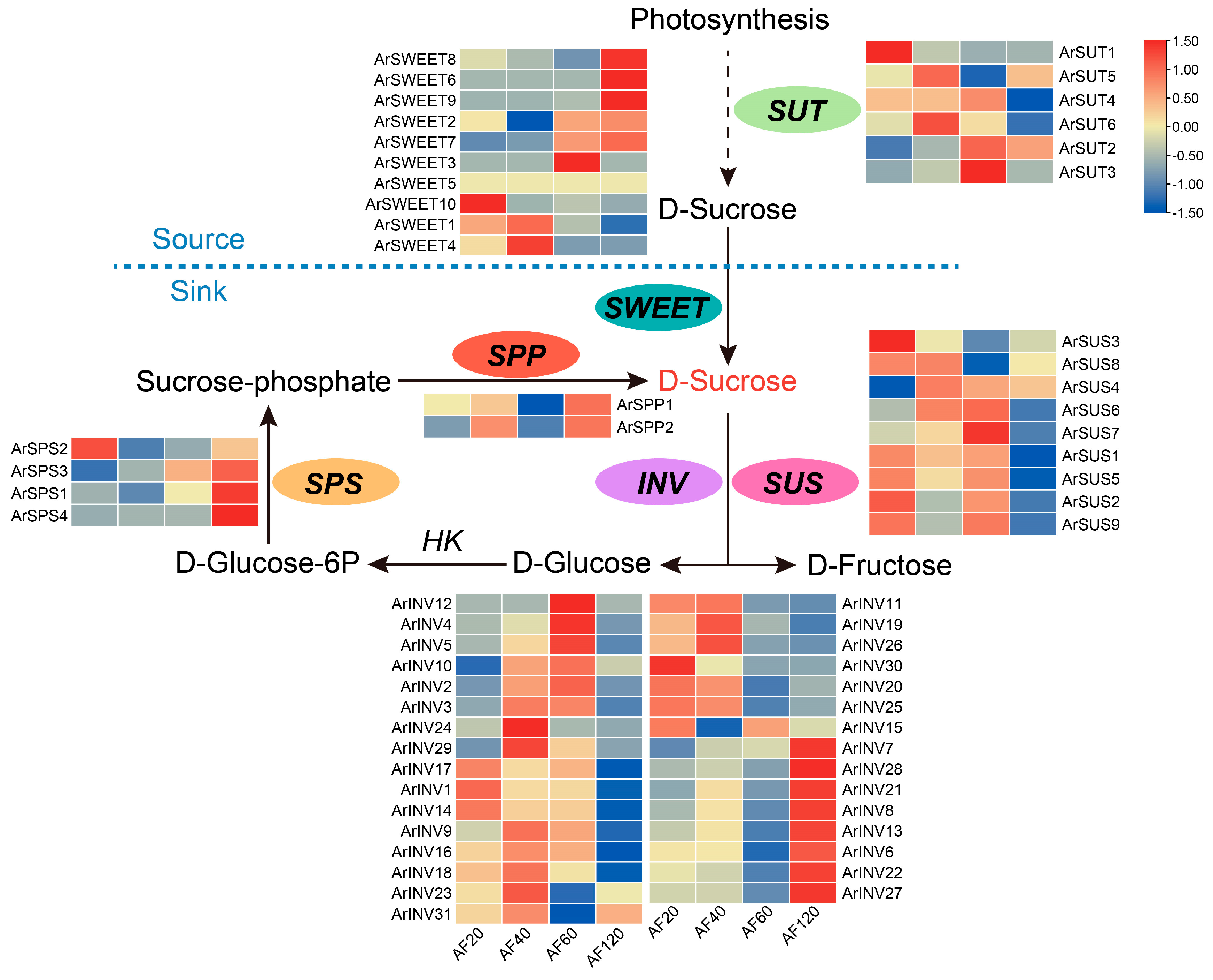
3.6. Genome-Wide Expression Analysis of Sucrose Metabolism Genes
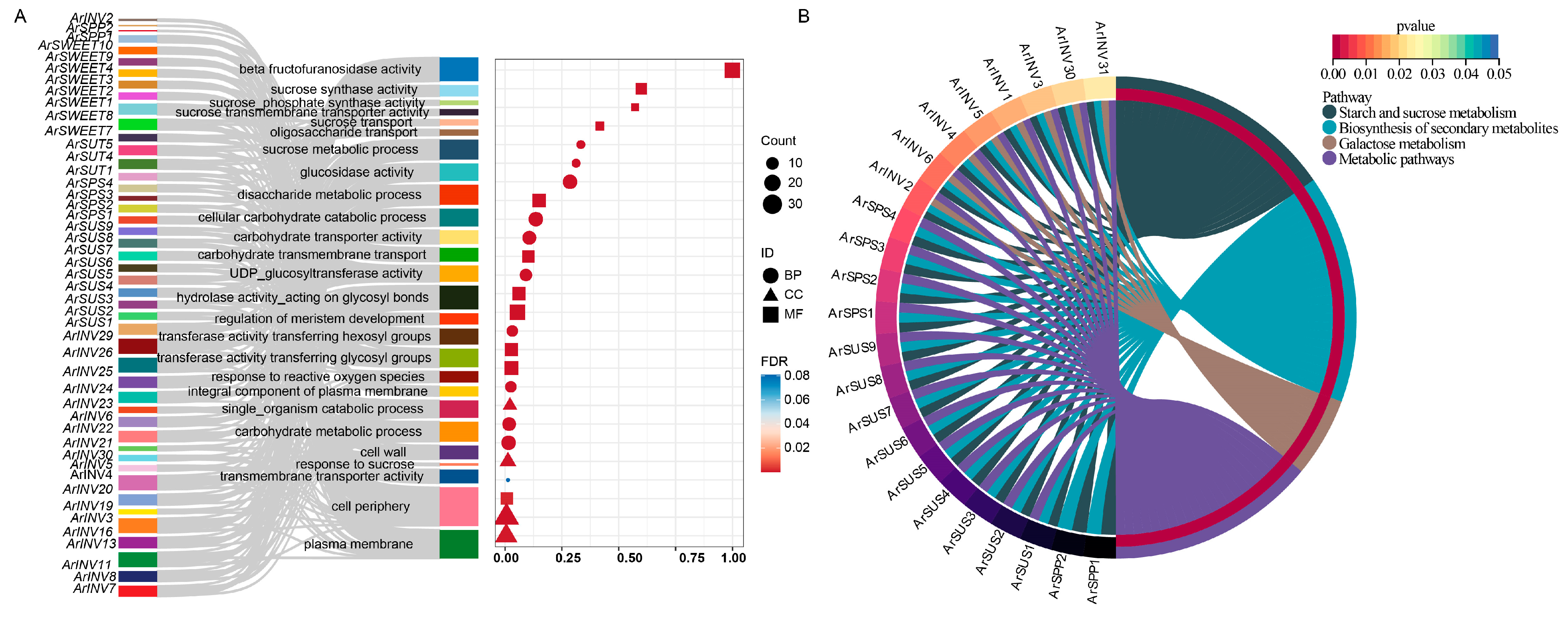
3.7. GO and KEGG Functional Annotation of Sucrose Metabolism Genes
3.8. Temporal Gene Expression Dynamics during Fruit Development and Ripening
3.9. Identification of Key TFs Regulating Sucrose Metabolism
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ruan, Y.L. Sucrose metabolism: Gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef] [PubMed]
- Rolland, F.; Baena-Gonzalez, E.; Sheen, J. Sugar sensing and signaling in plants: Conserved and novel mechanisms. Annu. Rev. Plant Biol. 2006, 57, 675–709. [Google Scholar] [CrossRef] [PubMed]
- Umer, M.J.; Bin, S.L.; Gebremeskel, H.; Zhao, S.; Yuan, P.; Zhu, H.; Kaseb, M.O.; Anees, M.; Lu, X.; He, N.; et al. Identification of key gene networks controlling organic acid and sugar metabolism during watermelon fruit development by integrating metabolic phenotypes and gene expression profiles. Hortic. Res. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ali, M.M.; Li, B.; Fang, T.; Chen, F. Transcriptome data-based identification of candidate genes involved in metabolism and accumulation of soluble sugars during fruit development in ‘Huangguan’ plum. J. Food Biochem. 2021, 45, e13878. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Quoc, B.; Foyer, C.H. A role for futile cycles involving invertase and sucrose synthase in sucrose metabolism of tomato fruit. J. Exp. Bot. 2001, 52, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Sugar metabolism as input signals and fuel for leaf senescence. Genes Genom. 2019, 41, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Van Bel, A. The phloem, a miracle of ingenuity. Plant Cell Environ. 2003, 26, 125–149. [Google Scholar] [CrossRef]
- Li, J.; Qin, M.; Qiao, X.; Cheng, Y.; Li, X.; Zhang, H.; Wu, J. A new insight into the evolution and functional divergence of SWEET transporters in chinese white pear (Pyrus bretschneideri). Plant Cell Physiol. 2017, 58, 839–850. [Google Scholar] [CrossRef]
- Xu, Y.; Yao, Z.; Cheng, Y.; Ruan, M.; Ye, Q.; Wang, R.; Zhou, G.; Liu, J.; Liu, C.; Wan, H. Divergent retention of sucrose metabolism genes after whole genome triplication in the tomato (Solanum lycopersicum). Plants 2023, 12, 4145. [Google Scholar] [CrossRef]
- Reuscher, S.; Akiyama, M.; Yasuda, T.; Makino, H.; Aoki, K.; Shibata, D.; Shiratake, K. The sugar transporter inventory of tomato: Genome-wide identification and expression analysis. Plant Cell Physiol. 2014, 55, 1123–1141. [Google Scholar] [CrossRef]
- Chen, L.Q.; Hou, B.H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.Q.; Guo, W.J.; Kim, J.G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef]
- Johnson, D.A.; Thomas, M.A. The monosaccharide transporter gene family in Arabidopsis and rice: A history of duplications, adaptive evolution, and functional divergence. Mol. Biol. Evol. 2007, 24, 2412–2423. [Google Scholar] [CrossRef]
- Fakher, B.; Jakada, B.H.; Greaves, J.G.; Wang, L.; Niu, X.; Cheng, Y.; Zheng, P.; Aslam, M.; Qin, Y.; Wang, X. Identification and expression analysis of pineapple sugar transporters reveal their role in the development and environmental response. Front. Plant Sci. 2022, 13, 964897. [Google Scholar] [CrossRef]
- Jiang, C.; Zeng, S.; Yang, J.; Wang, X. Genome-wide identification and expression profiling analysis of SWEET family genes involved in fruit development in plum (Prunus salicina Lindl). Genes 2023, 14, 1679. [Google Scholar]
- Wu, S.; Wu, D.; Song, J.; Zhang, Y.; Tan, Q.; Yang, T.; Yang, J.; Wang, S.; Xu, J.; Xu, W.; et al. Metabolomic and transcriptomic analyses reveal new insights into the role of abscisic acid in modulating mango fruit ripening. Hortic Res. 2022, 9, uhac102. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Shu, P.; Zhang, C.; Zhang, J.; Chen, Y.; Zhang, Y.; Du, K.; Xie, Y.; Li, M.; Ma, T.; et al. Integrative analyses of metabolome and genome-wide transcriptome reveal the regulatory network governing flavor formation in kiwifruit (Actinidia chinensis). New Phytol. 2022, 233, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Shu, P.; Zhang, Z.; Wu, Y.; Chen, Y.; Li, K.; Deng, H.; Zhang, J.; Zhang, X.; Wang, J.; Liu, Z.; et al. A comprehensive metabolic map reveals major quality regulations in red-flesh kiwifruit (Actinidia chinensis). New Phytol. 2023, 238, 2064–2079. [Google Scholar] [CrossRef]
- Kim, S.C.; Lee, J.W.; Baek, S.H.; Lee, M.W.; Kang, Y.J. The complete chloroplast genome sequence of Actinidia rufa (Actinidiaceae). Mitochondrial DNA B Resour. 2018, 3, 564–565. [Google Scholar] [CrossRef]
- Han, F.; Zhao, T.T.; Liu, X.L.; Zhang, Q.; Li, D.W.; Tian, H.; Peng, J.; Zhong, C.H. Genetic analysis of fruit traits in Actinidia rufa (Siebold and Zuccarini) Planchon ex Miquel × Actinidia chinensis var. chinensis C. F. Liang kiwifruit hybrid population. Plant Sci. J. 2022, 40, 505–512. [Google Scholar]
- Kisaki, G.; Shimagami, T.; Matsudaira, K.; Tsugi, Y.; Moriguchi, K.; Nakashima, K.; Morimoto, T.; Gomi, K.; Ichimura, K.; Hamano, K.; et al. A kiwifruit cultivar crossbred with Actinidia chinensis and Actinidia rufa has practical tolerance to Pseudomonas syringae pv. actinidiae biovar 3. J. Plant Pathol. 2019, 101, 1211–1214. [Google Scholar] [CrossRef]
- Huang, C.S.; Ma, S.Y.; Liu, H.X.; Lu, Q.; Shi, L.G.; Liao, N.; Wei, L.B. Chemical constituents from roots of Actinidia rufa and their cytotoxicity. Chin. J. Chin. Mater. Med. 2017, 42, 2714–2718. [Google Scholar]
- Liao, G.; Chen, L.; He, Y.; Li, X.; Lv, Z.; Yi, S.; Zhong, M.; Huang, C.; Jia, D.; Qu, X.; et al. Three metabolic pathways are responsible for the accumulation and maintenance of high AsA content in kiwifruit (Actinidia eriantha). BMC Genom. 2021, 22, 13. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, K.; Qi, Y.; Lv, G.; Ren, X.; Liu, Z.; Ma, F. Transcriptional regulation of anthocyanin synthesis by MYB-bHLH-WDR complexes in kiwifruit Actinidia chinensis. J. Agric. Food Chem. 2021, 69, 3677–3691. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, R.; Bulley, S.M.; Zhong, C.; Li, D. Kiwifruit MYBS1-like and GBF3 transcription factors influence L-ascorbic acid biosynthesis by activating transcription of GDP-L-galactose phosphorylase 3. New Phytol. 2022, 234, 1782. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xie, X.; Zhong, C.; Li, D. Comparative transcriptome analysis revealed the key genes regulating ascorbic acid synthesis in Actinidia. Int. J. Mol. Sci. 2021, 22, 12894. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liao, G.; Huang, C.; Zhong, M.; Jia, D.; Qu, X.; Liu, Q.; He, Y.; Li, Y. Differences of sucrose accumulation concentration and related genes expression between two sucrose accumulation types of Actinidia eriantha. Sci. Rep. 2020, 10, 20474. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Li, Y.; Wang, H.; Liu, Q.; Zhong, M.; Jia, D.; Huang, C.; Xu, X. Genome-wide identification and expression profiling analysis of sucrose synthase (SUS) and sucrose phosphate synthase (SPS) genes family in Actinidia chinensis and A. eriantha. BMC Plant Biol. 2022, 22, 215. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yang, Y.; Liu, C.; Sun, Y.; Zhang, T.; Hou, M.; Huang, S.; Yuan, H. The evolutionary history of the sucrose synthase gene family in higher plants. BMC Plant Biol. 2019, 19, 566. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Varkonyi-Gasic, E.; Shirasawa, K.; Catanach, A.; Henry, I.M.; Mertten, D.; Datson, P.; Masuda, K.; Fujita, N.; Kuwada, E.; et al. Recurrent neo-sex chromosome evolution in kiwifruit. Nat. Plants 2023, 9, 393–402. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinf. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible read trimming tool for Illumina NGS data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Putri, G.H.; Anders, S.; Pyl, P.T.; Pimanda, J.E.; Zanini, F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics 2022, 38, 2943–2945. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, X.; Tang, B.; Li, C.; Kou, Z.; Li, L.; Liu, W.; Wu, Y.; Kou, X.; Li, J.; et al. Protein Expression Landscape of Mouse Embryos during Pre-implantation Development. Cell Rep. 2017, 21, 3957–3969. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.L.; Jin, Y.; Yang, Y.J.; Li, G.J.; Boyer, J.S. Sugar input, metabolism, and signaling mediated by invertase: Roles in development, yield potential, and response to drought and heat. Mol. Plant 2010, 3, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.C.O.; Rocha, D.S.J.; Silva, G.C.B.; Santos, M.G.M.; Camillo, L.R.; de Oliveira, P.H.G.A.; Cavalari, A.A.; Costa, M.G.C. Dynamics of the sucrose metabolism and related gene expression in tomato fruits under water deficit. Physiol. Mol. Biol. Plants 2023, 29, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Feng, F.; Cheng, L. Expression Patterns of Genes Involved in Sugar Metabolism and Accumulation during Apple Fruit Development. PLoS ONE 2012, 7, e33055. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.Y.; Chi, Y.H.; Wang, J.Z.; Zhou, J.X.; Cheng, Y.S.; Zhang, B.L.; Ma, A.; Vanitha, J.; Ramachandran, S. Sucrose metabolism gene families and their biological functions. Sci. Rep. 2015, 5, 17583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, S.; Ren, Y.; Gan, C.; Li, B.; Fan, Y.; Zhao, X.; Yuan, Z. Identification, analysis and gene cloning of the SWEET gene family provide insights into sugar transport in Pomegranate (Punica granatum). Int. J. Mol. Sci. 2022, 23, 2471. [Google Scholar] [CrossRef]
- Hu, W.; Hua, X.; Zhang, Q.; Wang, J.; Shen, Q.; Zhang, X.; Wang, K.; Yu, Q.; Lin, Y.R.; Ming, R.; et al. New insights into the evolution and functional divergence of the SWEET family in Saccharum based on comparative genomics. BMC Plant Biol. 2018, 18, 270. [Google Scholar] [CrossRef]
- Veillet, F.; Gaillard, C.; Coutos-Thevenot, P.; La Camera, S. Targeting the AtCWIN1 gene to explore the role of invertases in sucrose transport in roots and during Botrytis cinerea infection. Front. Plant Sci. 2016, 7, 1899. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yang, K.; Li, G.; Li, Y.; Gao, Z. Identification and expression analyses of invertase genes in Moso Bamboo reveal their potential drought stress functions. Front. Genet. 2021, 12, 696300. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, B.; Huang, H.; Huang, W.; Zhang, Z.; Wang, Q.; Luo, H.; An, B. Metabolomic and transcriptomic analyses provide insights into metabolic networks during cashew fruit development and ripening. Food Chem. 2023, 404 Pt B, 134765. [Google Scholar] [CrossRef]
- Zhou, D.; Shen, Y.; Zhou, P.; Fatima, M.; Lin, J.; Yue, J.; Zhang, X.; Chen, L.Y.; Ming, R. Papaya CpbHLH1/2 regulate carotenoid biosynthesis-related genes during papaya fruit ripening. Hortic Res. 2019, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kang, J.; Xie, Q.; Gong, J.; Shen, H.; Chen, Y.; Chen, G.; Hu, Z. The basic helix-loop-helix transcription factor bHLH95 affects fruit ripening and multiple metabolisms in tomato. J. Exp. Bot. 2020, 71, 6311–6327. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Chen, G.; Zhou, S.; Tu, Y.; Wang, Y.; Dong, T.; Hu, Z. A new tomato NAC (NAM/ATAF1/2/CUC2) transcription factor, SlNAC4, functions as a positive regulator of fruit ripening and carotenoid accumulation. Plant Cell Physiol. 2014, 55, 119–135. [Google Scholar] [CrossRef]
- Cao, H.; Chen, J.; Yue, M.; Xu, C.; Jian, W.; Liu, Y.; Song, B.; Gao, Y.; Cheng, Y.; Li, Z. Tomato transcriptional repressor MYB70 directly regulates ethylene-dependent fruit ripening. Plant J. 2020, 104, 1568–1581. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Family | INV | SPP | SPS | SUS | SUC/SUT | SWEET | Total |
|---|---|---|---|---|---|---|---|
| Arabidopsis thaliana | 33 | 2 | 6 | 6 | 9 | 17 | 73 |
| Actinidia chinensis | 31 | 3 | 6 | 10 | 10 | 29 | 89 |
| Actinidia eriantha | 28 | 2 | 6 | 9 | 9 | 28 | 82 |
| Actinidia hemsleyana | 29 | 2 | 6 | 7 | 7 | 19 | 70 |
| Actinidia rufa | 31 | 2 | 4 | 9 | 6 | 10 | 62 |
| Solanum lycopersicum | 19 | 2 | 4 | 6 | 2 | 25 | 58 |
| Vitis vinifera | 21 | 5 | 7 | 7 | 5 | 20 | 65 |
| Gene Name | Gene ID | Protein Length (aa) | Molecular Weight (kDa) | Isoelectric Point |
|---|---|---|---|---|
| ArINV1 | GFY84308.1 | 443 | 49.10 | 4.53 |
| ArINV2 | GFY97846.1 | 533 | 59.94 | 6.35 |
| ArINV3 | GFZ01189.1 | 867 | 96.97 | 5.23 |
| ArINV4 | GFZ04274.1 | 452 | 50.42 | 6.56 |
| ArINV5 | GFZ04278.1 | 456 | 51.59 | 9.22 |
| ArINV6 | GFZ08509.1 | 507 | 57.58 | 6.37 |
| ArINV7 | GFS29709.1 | 296 | 34.36 | 6.96 |
| ArINV8 | GFS29713.1 | 379 | 42.65 | 5.4 |
| ArINV9 | GFS39341.1 | 598 | 67.29 | 6.51 |
| ArINV10 | GFS42808.1 | 570 | 64.96 | 5.88 |
| ArINV11 | GFY85269.1 | 566 | 63.75 | 6.08 |
| ArINV12 | GFY86245.1 | 570 | 65.27 | 6.24 |
| ArINV13 | GFY89074.1 | 597 | 67.79 | 6.18 |
| ArINV14 | GFY89880.1 | 574 | 65.60 | 6.02 |
| ArINV15 | GFY90201.1 | 508 | 57.77 | 8.72 |
| ArINV16 | GFY95297.1 | 552 | 63.07 | 6.31 |
| ArINV17 | GFZ00601.1 | 576 | 65.73 | 6.08 |
| ArINV18 | GFZ00609.1 | 615 | 70.02 | 6.24 |
| ArINV19 | GFZ03044.1 | 191 | 21.67 | 5.24 |
| ArINV20 | GFZ03045.1 | 106 | 11.80 | 5.57 |
| ArINV21 | GFZ07245.1 | 645 | 73.37 | 5.53 |
| ArINV22 | GFZ07303.1 | 571 | 64.00 | 5.35 |
| ArINV23 | GFZ09104.1 | 466 | 52.63 | 5.74 |
| ArINV24 | GFZ09106.1 | 540 | 61.00 | 6.55 |
| ArINV25 | GFZ09762.1 | 105 | 12.04 | 9.36 |
| ArINV26 | GFZ09763.1 | 382 | 43.82 | 5.71 |
| ArINV27 | GFZ11195.1 | 654 | 73.46 | 5.88 |
| ArINV28 | GFZ12668.1 | 618 | 68.82 | 6.09 |
| ArINV29 | GFZ21641.1 | 668 | 75.08 | 6.31 |
| ArINV30 | GFZ06978.1 | 652 | 73.02 | 4.99 |
| ArINV31 | GFZ15206.1 | 514 | 58.10 | 6.64 |
| ArSPP1 | GFY98003.1 | 395 | 44.70 | 5.6 |
| ArSPP2 | GFZ09269.1 | 433 | 48.91 | 5.56 |
| ArSPS1 | GFS40808.1 | 716 | 80.76 | 6.05 |
| ArSPS2 | GFY88805.1 | 566 | 63.59 | 5.95 |
| ArSPS3 | GFY95144.1 | 983 | 112.32 | 8.64 |
| ArSPS4 | GFY99702.1 | 811 | 90.89 | 6.13 |
| ArSUS1 | GFS35293.1 | 559 | 63.66 | 6.51 |
| ArSUS2 | GFS35295.1 | 717 | 80.50 | 5.76 |
| ArSUS3 | GFS35628.1 | 815 | 93.07 | 5.7 |
| ArSUS4 | GFS35631.1 | 714 | 80.91 | 7.67 |
| ArSUS5 | GFS37905.1 | 838 | 94.50 | 8.13 |
| ArSUS6 | GFS38485.1 | 806 | 92.63 | 5.97 |
| ArSUS7 | GFY98571.1 | 772 | 88.84 | 6.13 |
| ArSUS8 | GFZ09599.1 | 1240 | 140.46 | 9.09 |
| ArSUS9 | GFZ17994.1 | 819 | 93.16 | 8.07 |
| ArSUT1 | GFS44113.1 | 452 | 49.42 | 9.48 |
| ArSUT2 | GFY85479.1 | 493 | 53.53 | 9.28 |
| ArSUT3 | GFY85509.1 | 518 | 54.68 | 9.17 |
| ArSUT4 | GFY85510.1 | 499 | 53.00 | 9 |
| ArSUT5 | GFY88434.1 | 591 | 63.84 | 6.8 |
| ArSUT6 | GFY96916.1 | 504 | 53.61 | 8.94 |
| ArSWEET1 | GFS37554.1 | 210 | 23.00 | 9.41 |
| ArSWEET2 | GFS38071.1 | 280 | 30.70 | 7.76 |
| ArSWEET3 | GFS38076.1 | 169 | 18.33 | 9.55 |
| ArSWEET4 | GFS45098.1 | 69 | 7.68 | 8.01 |
| ArSWEET5 | GFY89003.1 | 210 | 23.25 | 6.5 |
| ArSWEET6 | GFY99936.1 | 231 | 25.82 | 6.95 |
| ArSWEET7 | GFZ03923.1 | 247 | 27.40 | 9.37 |
| ArSWEET8 | GFZ03924.1 | 220 | 25.07 | 8.58 |
| ArSWEET9 | GFZ04631.1 | 195 | 22.06 | 5.6 |
| ArSWEET10 | GFZ09897.1 | 252 | 27.56 | 9.71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Y.; Qiang, X.; Dong, P.; Ren, T.; Zhang, Y.; Yang, Y. Genome-Wide Identification and Characterization of Sucrose Metabolism Genes Involved in Actinidia rufa and Their Expression Profiling during the Fruit Developmental Stages. Horticulturae 2024, 10, 772. https://doi.org/10.3390/horticulturae10070772
Jia Y, Qiang X, Dong P, Ren T, Zhang Y, Yang Y. Genome-Wide Identification and Characterization of Sucrose Metabolism Genes Involved in Actinidia rufa and Their Expression Profiling during the Fruit Developmental Stages. Horticulturae. 2024; 10(7):772. https://doi.org/10.3390/horticulturae10070772
Chicago/Turabian StyleJia, Yun, Xu Qiang, Pengbin Dong, Ting Ren, Ying Zhang, and Yanci Yang. 2024. "Genome-Wide Identification and Characterization of Sucrose Metabolism Genes Involved in Actinidia rufa and Their Expression Profiling during the Fruit Developmental Stages" Horticulturae 10, no. 7: 772. https://doi.org/10.3390/horticulturae10070772