
C-CorA: A Cluster-Based Method for Correlation Analysis of RNA-Seq Data



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. RNA-Seq Analysis
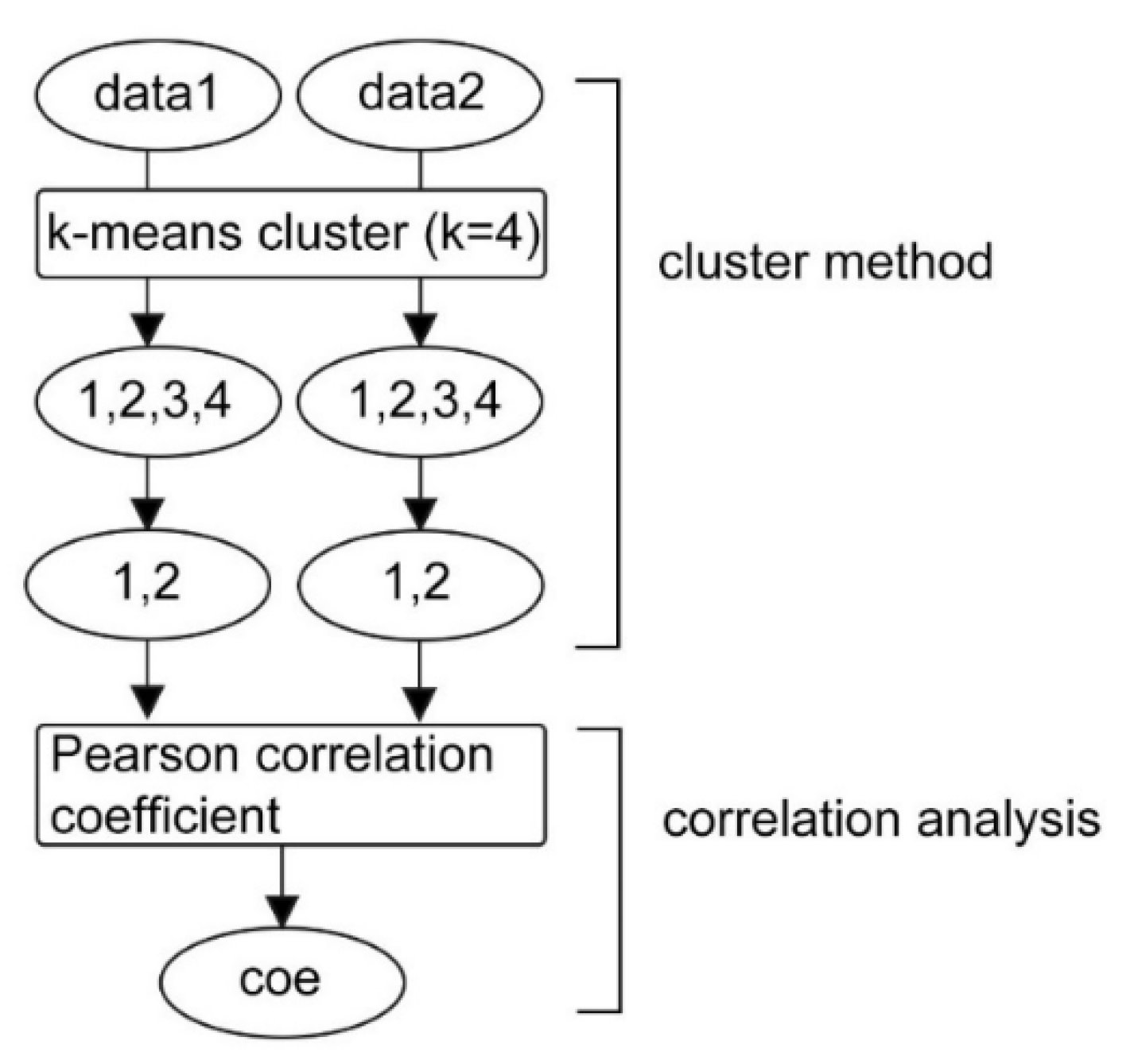
2.3. Cluster-Based Correlation Method (C-CorA)
| Algorithm 1: Cluster-Based Correlation Coefficient Calculation. |
| Input: Spheno, Sexp, k, p Output: Coe 1: 2: fori = 1, 2, …, 7 do 3: 4: end for 5: while readline Sexp do 6: 7: for i = 1, 2, …, 7 do 8: 9: for j = 1, 2, …, 7 do 10: c = abs(corr(), pearson) 11: if c ≥ p then 12: store c in Coe 13: end if 14: end for 15: end for 16: end while |
3. Results
3.1. Method Implementation
3.2. Transcriptome Analysis Using C-CorA Method
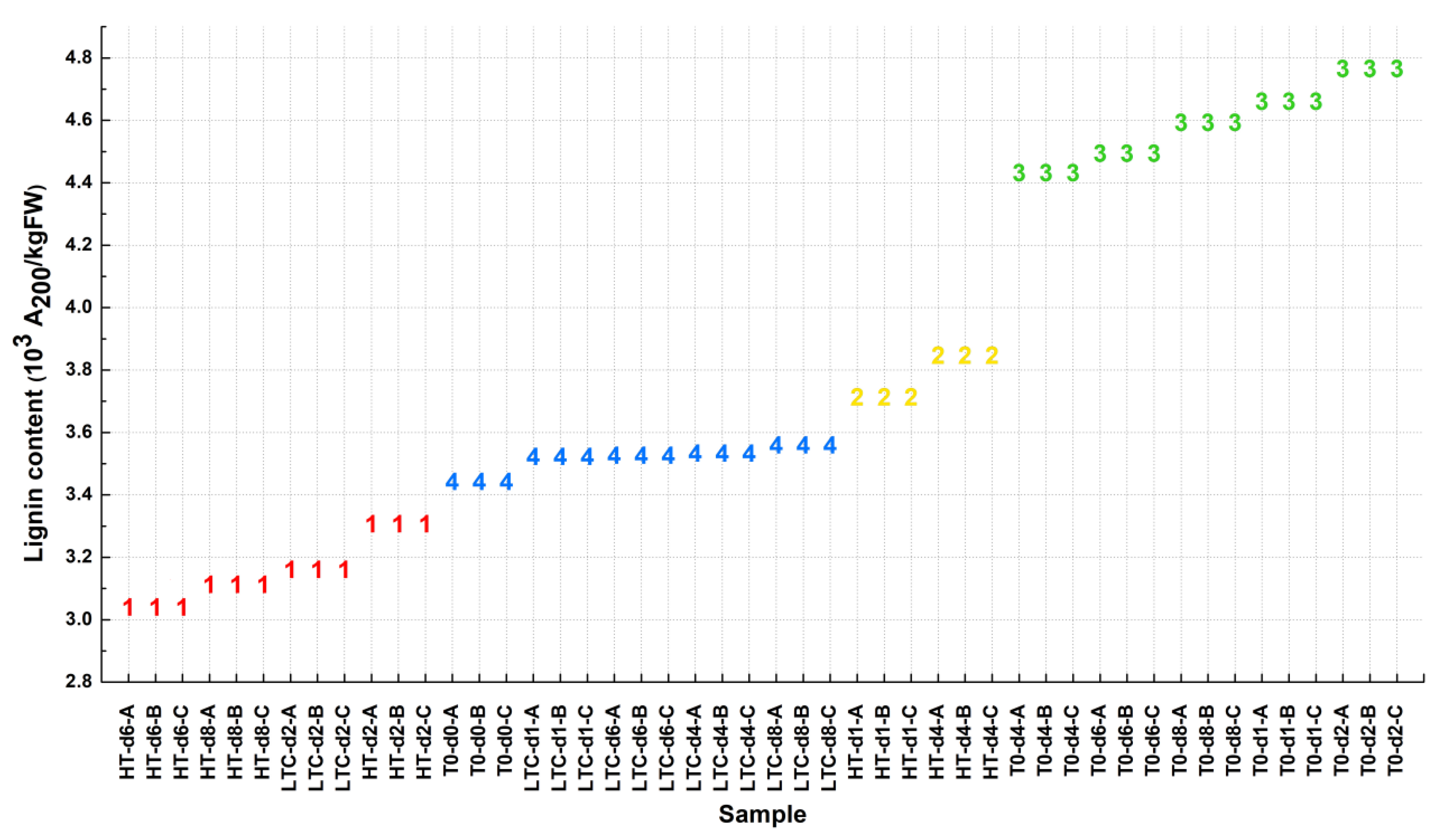
3.2.1. Clustering of Loquat Samples Based on Lignin Content
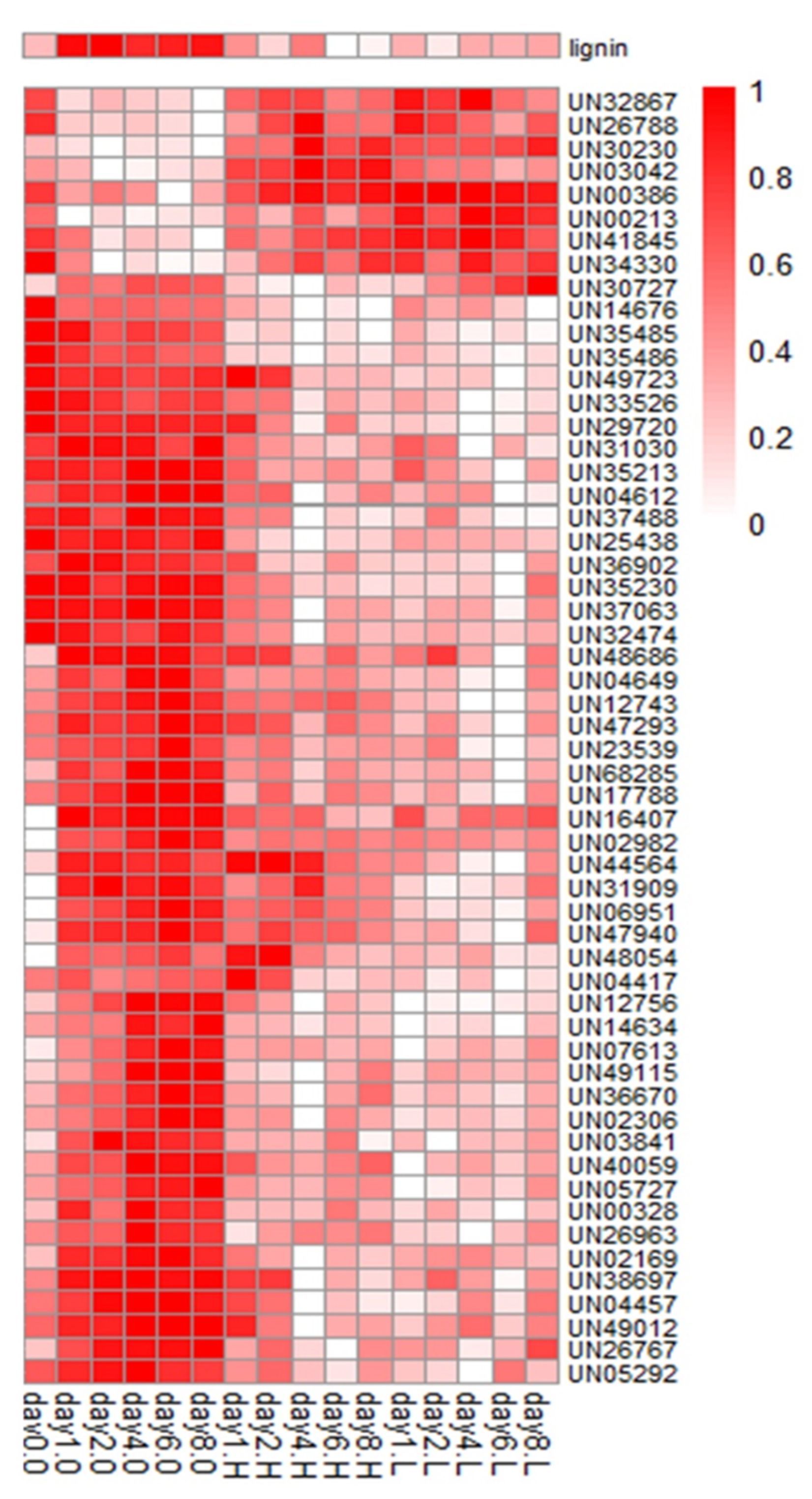
3.2.2. Clustering of Gene Expression Data
3.2.3. Correlation Analysis of Gene Expression vs. Lignin Content
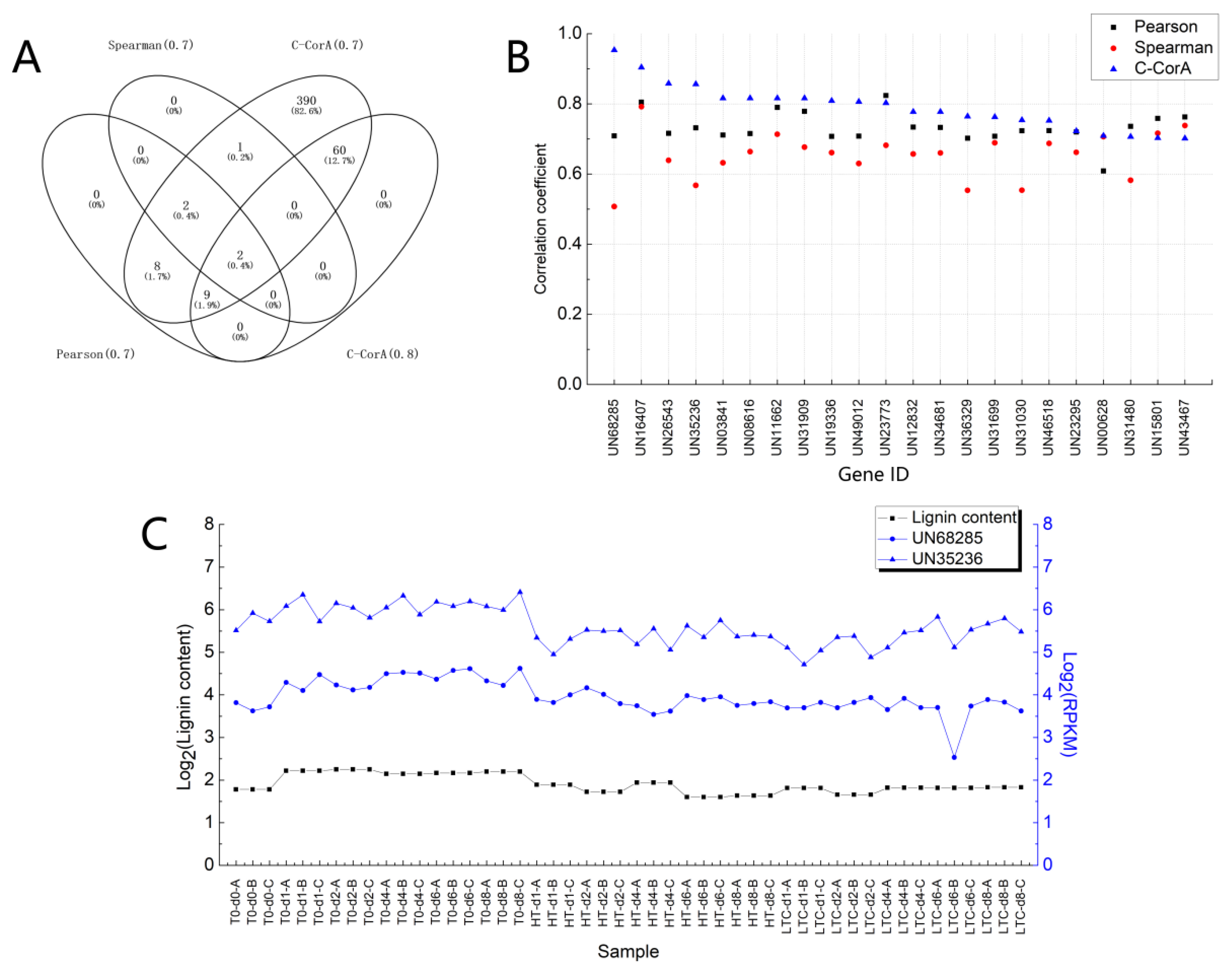
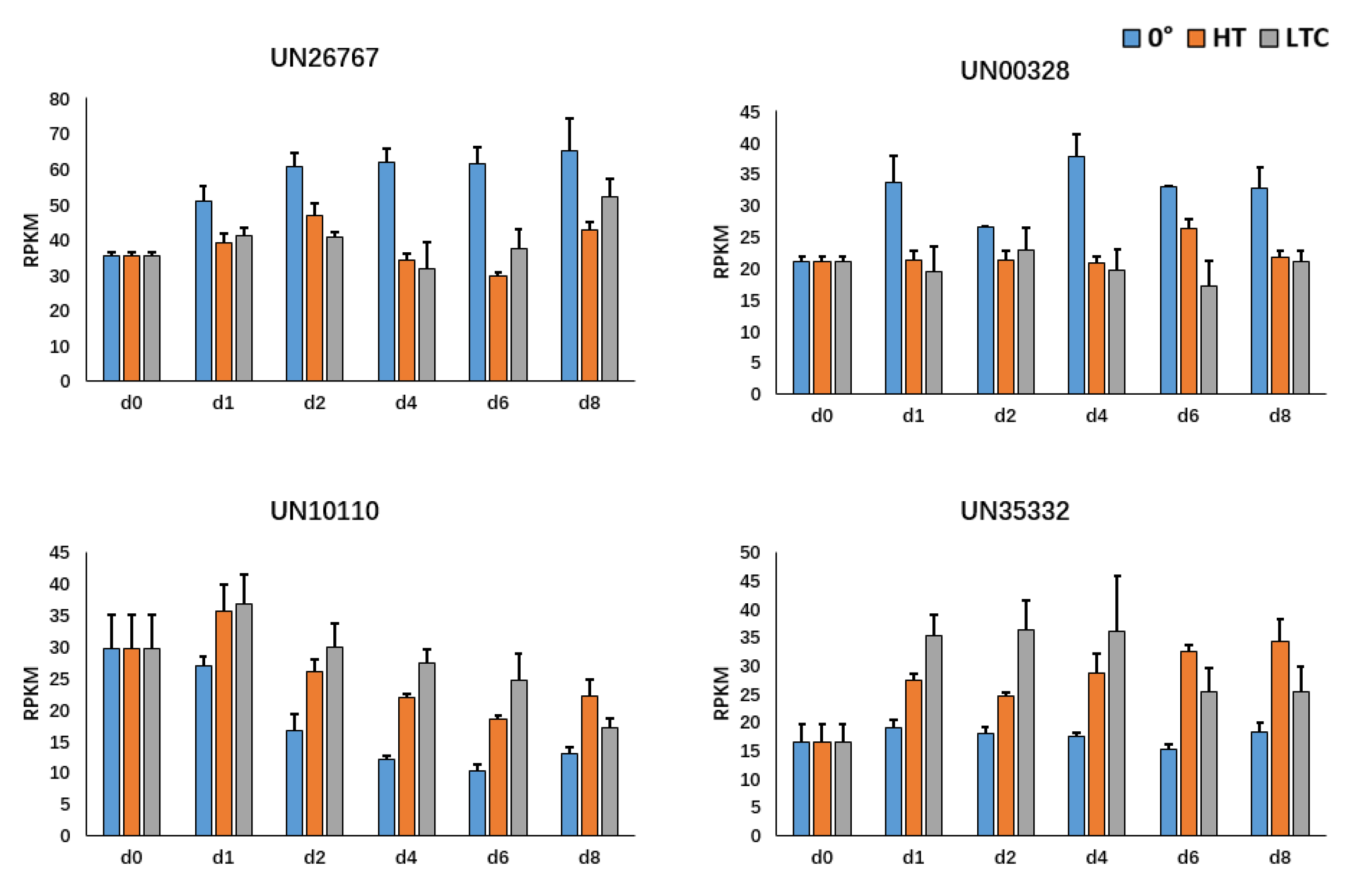
3.2.4. Correlation Analysis for Newly Identified Candidate Genes
3.3. Other Cases of Correlation Analysis Using C-CorA
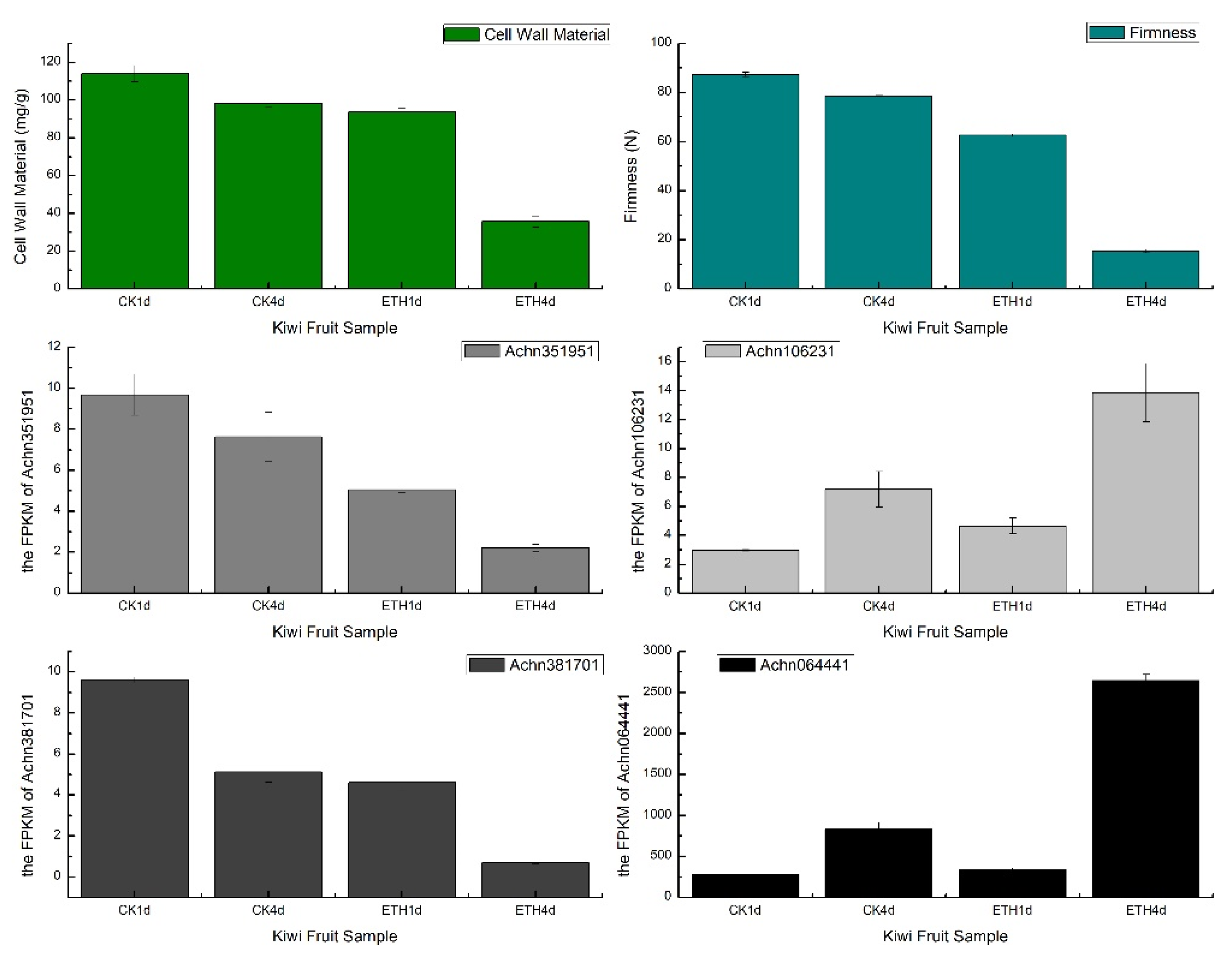
3.3.1. Transcriptome Analysis in Kiwifruit Using C-CorA
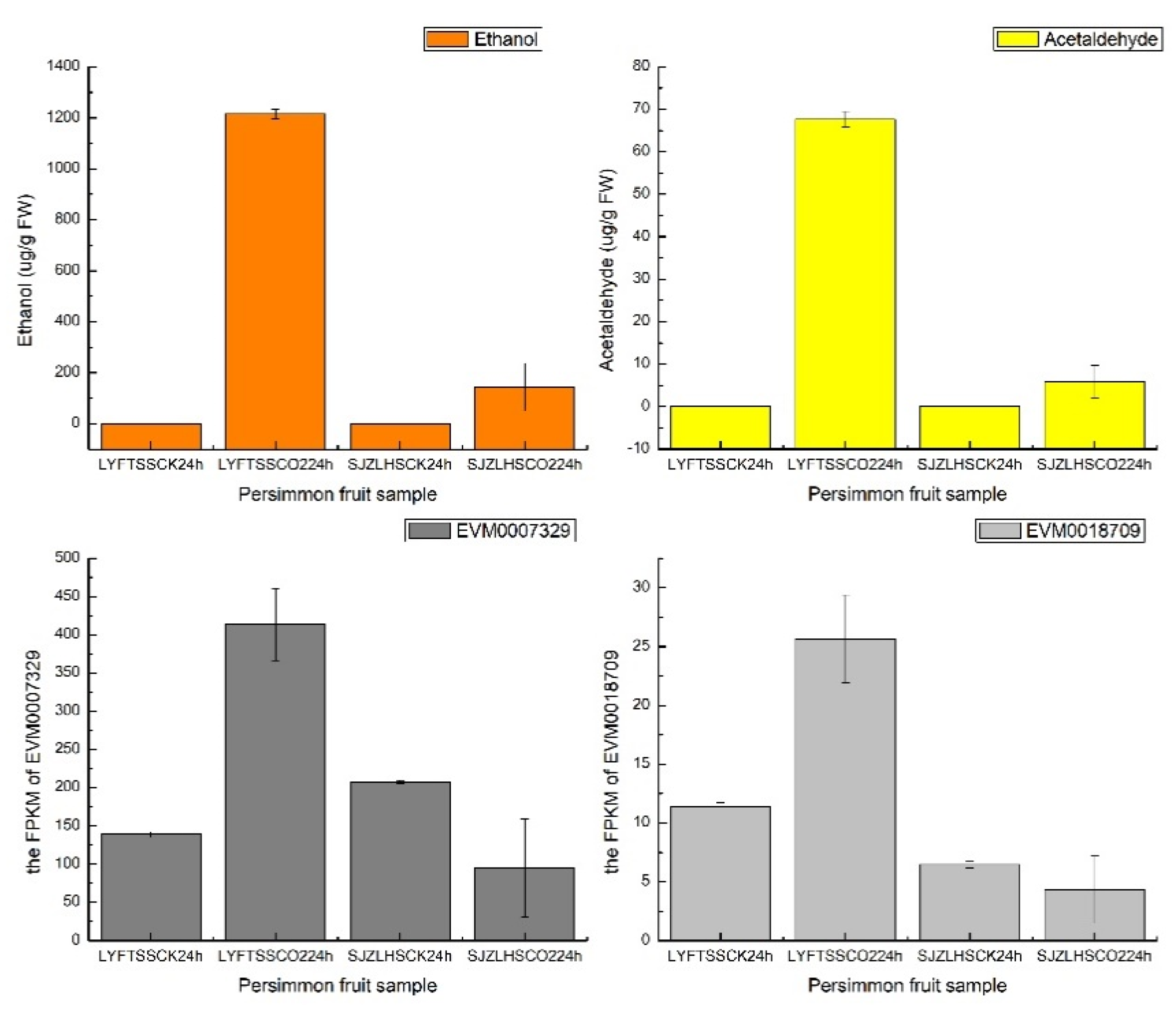
3.3.2. Transcriptome Analysis in Persimmon Using C-CorA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, Y.; Zhan, M. Exploring pathways from gene co-expression to network dynamics. In Computational Systems Biology. Methods in Molecular Biology; Ireton, R., Montgomery, K., Bumgarner, R., Samudrala, R., McDermott, J., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 249–267. [Google Scholar]
- Sax, K.; Gowen, J.W. Permanence of tree performance in a clonal variety and a critique of the theory of bud mutation. Genetics 1923, 8, 179. [Google Scholar] [CrossRef]
- Cai, C.; Chen, K.; Xu, W.; Zhang, W.; Li, X.; Ferguson, I. Effect of 1-MCP on postharvest quality of loquat fruit. Postharvest Biol. Technol. 2006, 40, 155–162. [Google Scholar] [CrossRef]
- Cai, C.; Xu, C.; Li, X.; Ferguson, I.; Chen, K. Accumulation of lignin in relation to change in activities of lignification enzymes in loquat fruit flesh after harvest. Postharvest Biol. Technol. 2006, 40, 163–169. [Google Scholar] [CrossRef]
- Xu, Q.; Yin, X.R.; Zeng, J.K.; Ge, H.; Song, M.; Xu, C.J.; Chen, K.S. Activator-and repressor-type MYB transcription factors are involved in chilling injury induced flesh lignification in loquat via their interactions with the phenylpropanoid pathway. J. Exp. Bot. 2014, 65, 4349–4359. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Q.; Zhang, J.; Ge, H.; Li, S.J.; Li, X.; Yin, X.R.; Chen, K.S. EjMYB8 transcriptionally regulates flesh lignification in loquat fruit. PLoS ONE 2016, 11, e0154399. [Google Scholar] [CrossRef]
- Zeng, J.K.; Li, X.; Xu, Q.; Chen, J.Y.; Yin, X.R.; Ferguson, I.B.; Chen, K.S. EjAP2-1, an AP 2/ERF gene, is a novel regulator of fruit lignification induced by chilling injury, via interaction with Ej MYB transcription factors. Plant Biotechnol. J. 2015, 13, 1325–1334. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, J.; Jiao, C.; Yin, X.; Fei, Z.; Wu, Q.; Chen, K. Transcriptome analysis provides insights into the regulation of metabolic processes during postharvest cold storage of loquat (Eriobotrya japonica) fruit. Hortic. Res. 2019, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.L.; Li, X.; Wang, P.; Cai, C.; Zhang, B.; De Sun, C.; Chen, K.S. Characterization of cDNAs associated with lignification and their expression profiles in loquat fruit with different lignin accumulation. Planta 2008, 227, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Joung, J.G.; Zheng, Y.; Chen, Y.R.; Liu, B.; Shao, Y.; Giovannoni, J.J. High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harbor Protoc. 2011, 2011, 940. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Regev, A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Zhao, L.; Gao, J.; Fei, Z. iAssembler: A package for de novo assembly of Roche-454/Sanger transcriptome sequences. BMC Bioinform. 2011, 12, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Mo, H.; Im Kim, J.; Chapple, C. Genetic engineering of Arabidopsis to overproduce disinapoyl esters, potential lignin modification molecules. Biotechnol. Biofuels 2017, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tronchet, M.; Balague, C.; Kroj, T.; Jouanin, L.; Roby, D. Cinnamyl alcohol dehydrogenases-C and D, key enzymes in lignin biosynthesis, play an essential role in disease resistance in Arabidopsis. Mol. Plant Pathol. 2010, 11, 83–92. [Google Scholar] [CrossRef]
- Du, Q.; Gong, C.; Pan, W.; Zhang, D. Development and application of microsatellites in candidate genes related to wood properties in the Chinese white poplar (Populus tomentosa Carr.). DNA Res. 2013, 20, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Knight, H.; Trewavas, A.J.; Knight, M.R. Cold calcium signaling in Arabidopsis involves two cellular pools and a change in calcium signature after acclimation. Plant Cell 1996, 8, 489–503. [Google Scholar] [PubMed] [Green Version]
- Lu, G.; Li, Z.; Zhang, X.; Wang, R.; Yang, S. Expression analysis of lignin-associated genes in hard end pear (Pyrus pyrifolia Whangkeumbae) and its response to calcium chloride treatment conditions. J. Plant Growth Regul. 2015, 34, 251–262. [Google Scholar] [CrossRef]
- Figueroa, C.R.; Opazo, M.C.; Vera, P.; Arriagada, O.; Díaz, M.; Moya-León, M.A. Effect of postharvest treatment of calcium and auxin on cell wall composition and expression of cell wall-modifying genes in the Chilean strawberry (Fragaria chiloensis) fruit. Food Chem. 2012, 132, 2014–2022. [Google Scholar] [CrossRef]
- Braccini, I.; Pérez, S. Molecular basis of Ca2+-induced gelation in alginates and pectins: The egg-box model revisited. Biomacromolecules 2001, 2, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Ge, J.; Liu, X.C.; Wang, W.Q.; Liu, X.F.; Yin, X.R. Consensus Co-expression network analysis identifies AdZAT5 regulating pectin degradation in ripening kiwifruit. J. Adv. Res. 2021; in press. [Google Scholar] [CrossRef]
- Anderson, C.T. We be jammin’: An update on pectin biosynthesis, trafficking and dynamics. J. Exp. Bot. 2015, 67, erv501. [Google Scholar] [CrossRef] [Green Version]
- Min, T.; Yin, X.R.; Shi, Y.N.; Luo, Z.R.; Yao, Y.C.; Grierson, D.; Ferguson, I.B.; Chen, K.S. Ethylene-responsive transcription factors interact with promoters of ADH and PDC involved in persimmon (Diospyros kaki) fruit de-astringency. J. Exp. Bot. 2012, 63, 6393–6405. [Google Scholar] [CrossRef] [Green Version]
- Kou, S.M.; Jin, R.; Wu, Y.Y.; Huang, J.W.; Zhang, Q.Y.; Sun, N.J.; Yang, Y.; Guan, C.F.; Wang, W.Q.; Zhu, C.Q.; et al. Transcriptome analysis revealed the roles of carbohydrate metabolism on differential acetaldehyde production capacity in persimmon fruit in response to high-CO2 treatment. J. Agric. Food Chem. 2021, 69, 836–845. [Google Scholar] [CrossRef]
- Nicolas, B.; Harold, P.; Páll, M.; Lior, P. Near-optimal RNA-Seq quantification. Nat. Biotechnol. 2015, 34, 525–527. [Google Scholar]
- Patro, R.; Mount, S.M.; Kingsford, C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 2014, 32, 462–464. [Google Scholar] [CrossRef] [Green Version]
- Salentijn, E.M.; Aharoni, A.; Schaart, J.G.; Boone, M.J.; Krens, F.A. Differential gene expression analysis of strawberry cultivars that differ in fruit-firmness. Physiol. Plant. 2003, 118, 571–578. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, M.X.; Shi, Y.N.; Liu, X.F.; Li, X.; Grierson, D.; Chen, K.S. EjHAT1 participates in heat alleviation of loquat fruit lignification by suppressing the promoter activity of key lignin monomer synthesis gene EjCAD5. J. Agric. Food Chem. 2019, 67, 5204–5211. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2-Group Pattern | Correlation Coefficient | ||
|---|---|---|---|
| 0.7 | 0.8 | 0.9 | |
| [{1}, {2, 3, 4}] | 20 | 1 | 0 |
| [{4}, {1, 2, 3}] | 445 | 76 | 3 |
| [{2}, {1, 3, 4}] | 51 | 9 | 1 |
| [{3}, {1, 2, 4}] | 472 | 71 | 10 |
| [{1, 4}, {2, 3}] | 230 | 9 | 0 |
| [{1, 2}, {3, 4}] | 93 | 28 | 0 |
| [{1, 3}, {2, 4}] | 40 | 2 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, J.; Liu, W.; Shi, Y.; Zhang, M.; Wu, Q.; Chen, K.; Chen, W. C-CorA: A Cluster-Based Method for Correlation Analysis of RNA-Seq Data. Horticulturae 2022, 8, 124. https://doi.org/10.3390/horticulturae8020124
Qian J, Liu W, Shi Y, Zhang M, Wu Q, Chen K, Chen W. C-CorA: A Cluster-Based Method for Correlation Analysis of RNA-Seq Data. Horticulturae. 2022; 8(2):124. https://doi.org/10.3390/horticulturae8020124
Chicago/Turabian StyleQian, Jianpu, Wenli Liu, Yanna Shi, Mengxue Zhang, Qingbiao Wu, Kunsong Chen, and Wenbo Chen. 2022. "C-CorA: A Cluster-Based Method for Correlation Analysis of RNA-Seq Data" Horticulturae 8, no. 2: 124. https://doi.org/10.3390/horticulturae8020124
APA StyleQian, J., Liu, W., Shi, Y., Zhang, M., Wu, Q., Chen, K., & Chen, W. (2022). C-CorA: A Cluster-Based Method for Correlation Analysis of RNA-Seq Data. Horticulturae, 8(2), 124. https://doi.org/10.3390/horticulturae8020124