
Comparative Transcriptomic Analysis Provides Insight into the Key Regulatory Pathways and Differentially Expressed Genes in Blueberry Flower Bud Endo- and Ecodormancy Release

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
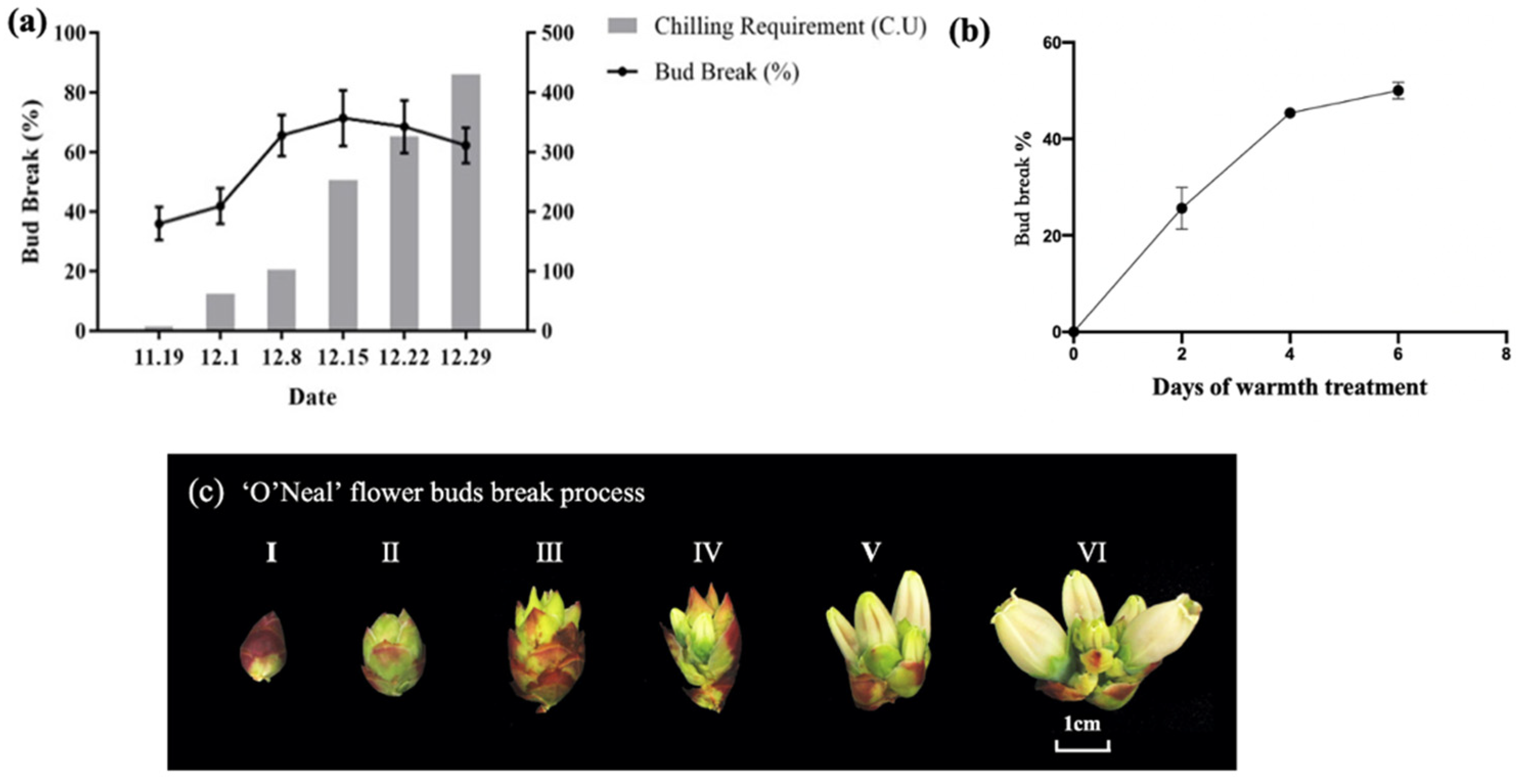
2.2. Identification of the Chilling Requirement and Dormancy Status of O’Neal
2.3. Artificial Warming Treatment
2.4. Preparation of the RNA-seq Library
2.5. Data Quality Control and Functional Annotation
2.6. Screening and Analysis of Differentially Expressed Genes
2.7. Hormone Profiling
2.8. WGCNA
2.9. Quantitative RT–PCR (qRT–PCR) Validation
2.10. Statistical Analysis
3. Results
3.1. Identification of the Dormancy Stage of Blueberry Flower Buds
3.2. RNA Sequencing Data Analysis
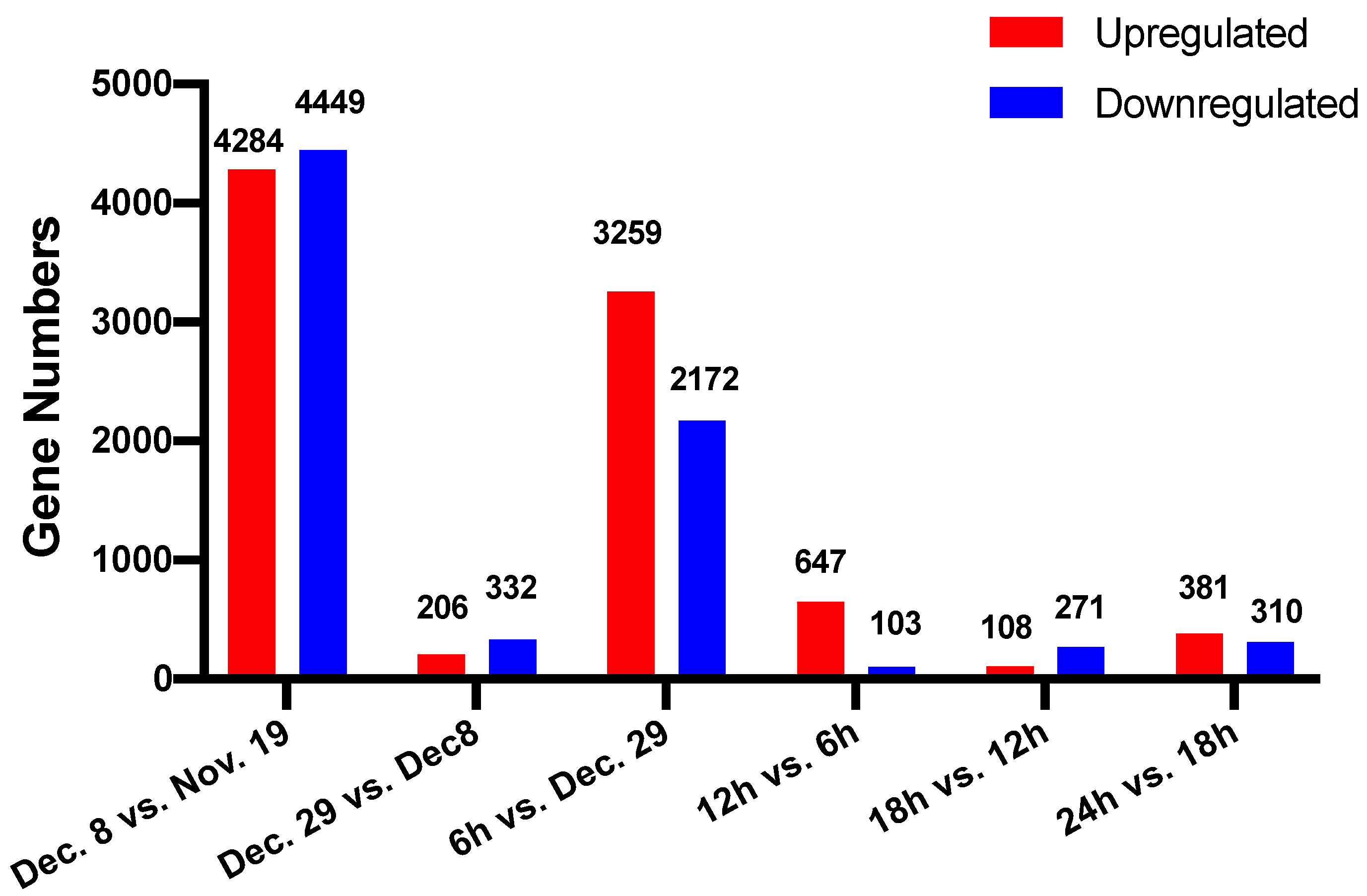
3.3. Screening of Differentially Expressed Genes during Dormancy Release
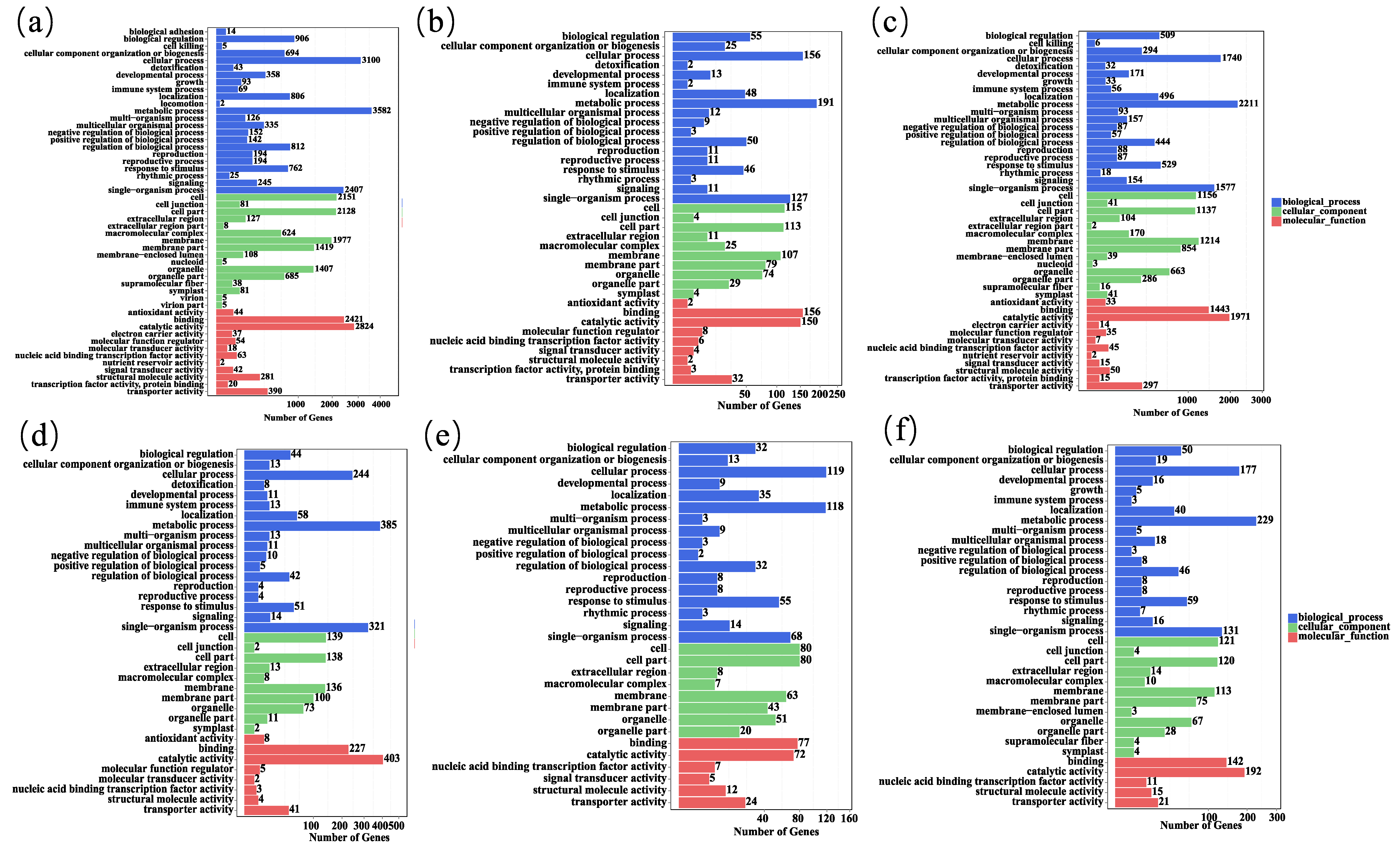
3.4. GO Enrichment Analysis of Differentially Expressed Genes
3.5. KEGG Enrichment Analysis of the DEGs
3.6. Analysis of Differentially Expressed Genes in Circadian Rhythm and Key Metabolic Pathways during Dormancy Release
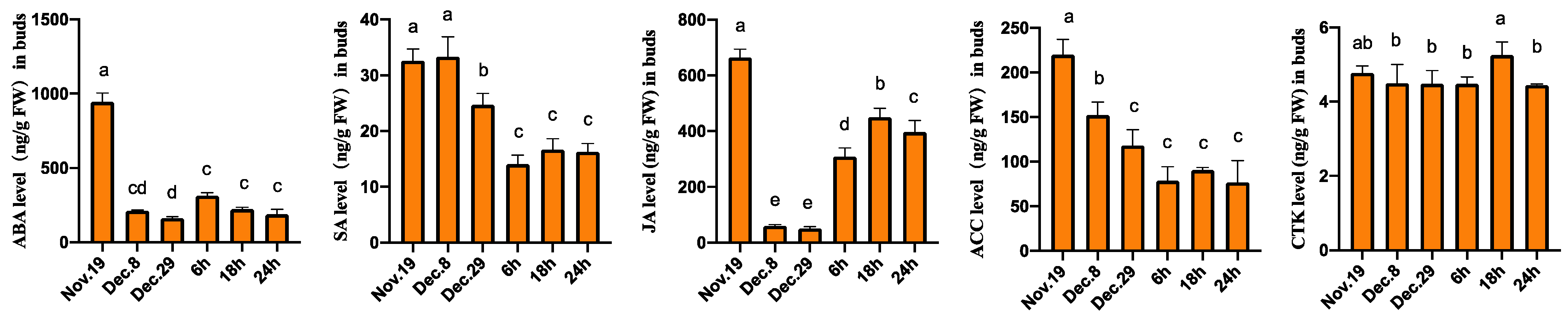
3.7. Changes in Hormone Levels during the Dormancy Release of Flower Buds
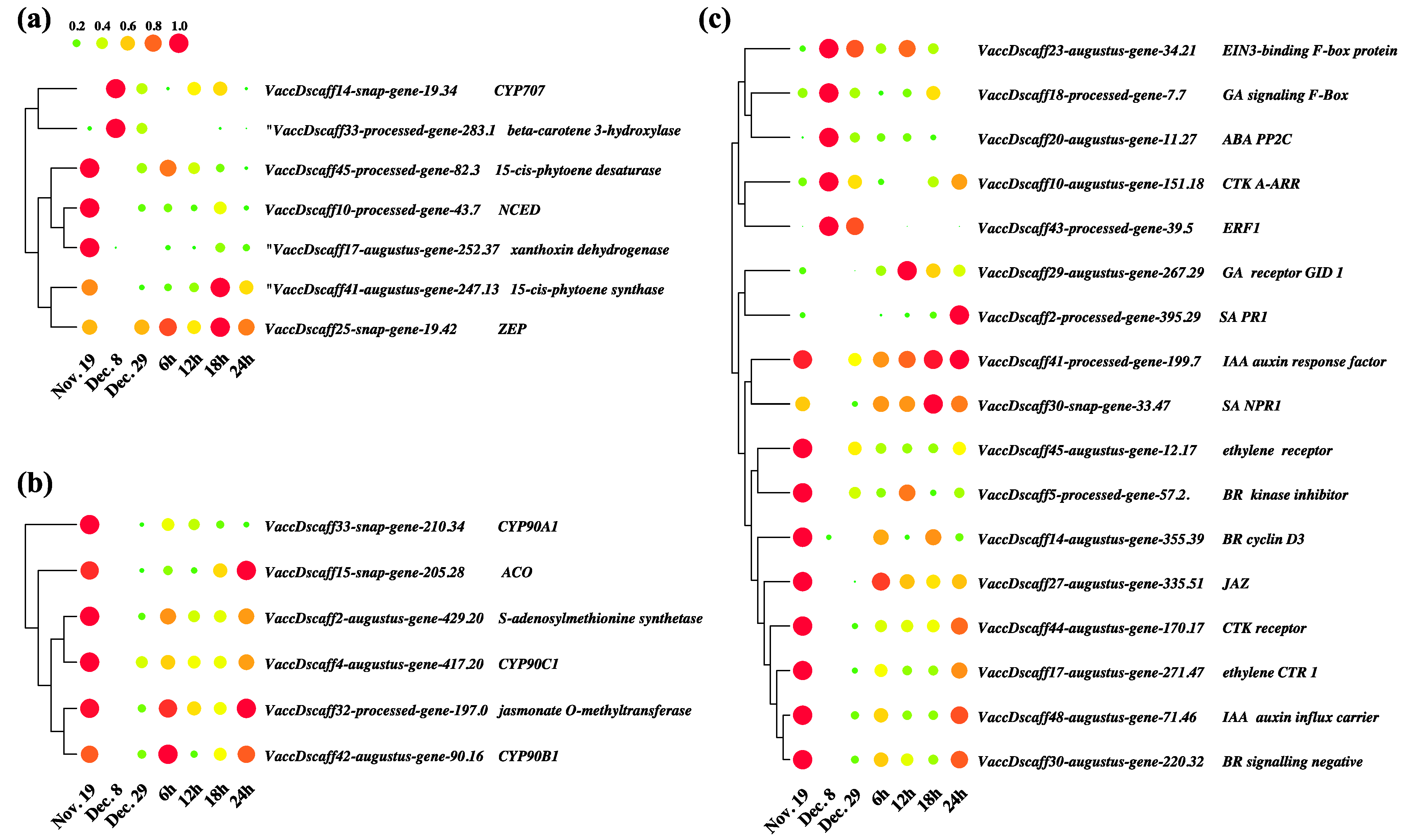
3.8. Key Differentially Expressed Genes in Hormone Anabolism and Signal Transduction Pathways
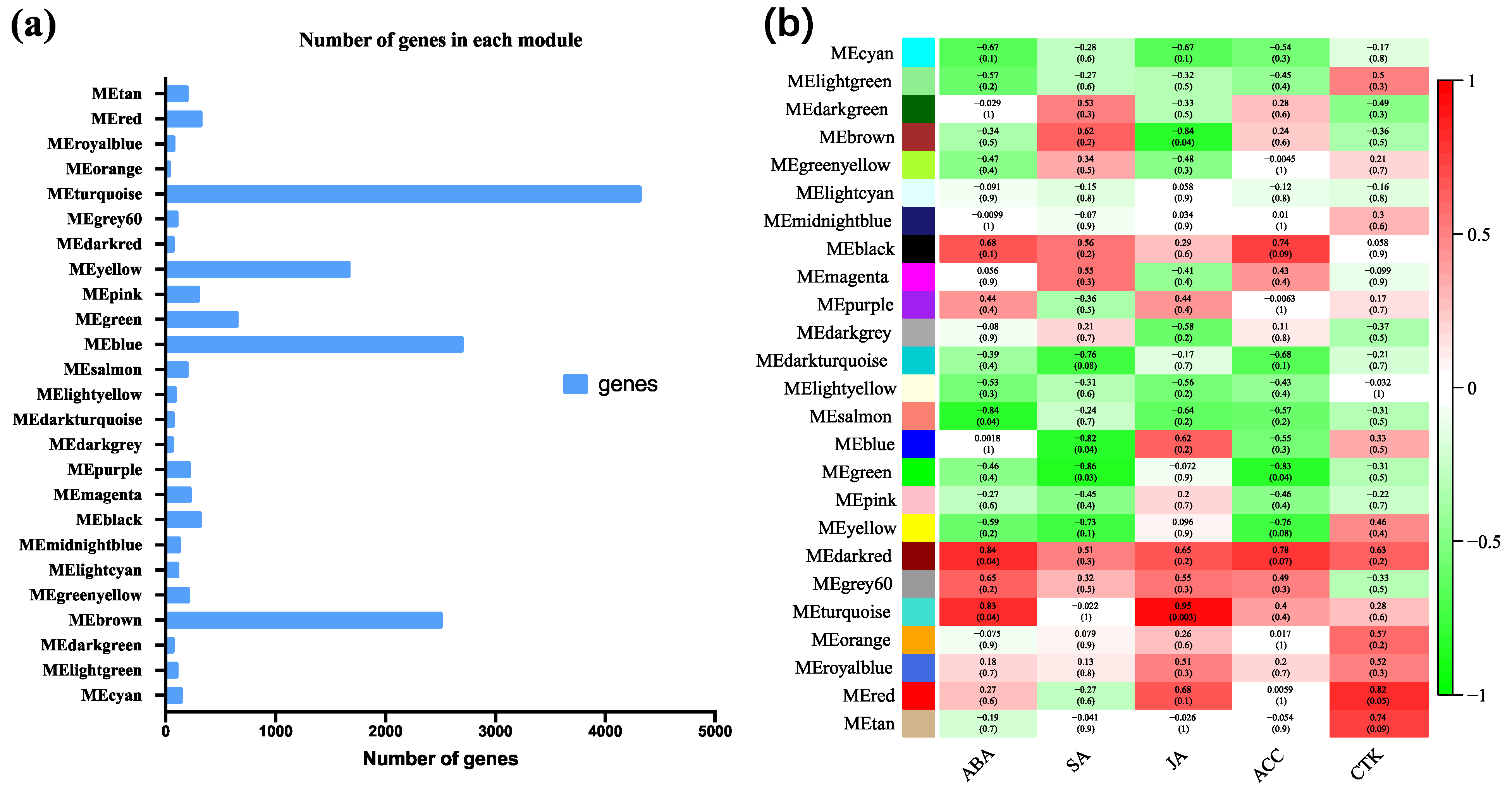
3.9. Coexpression Analysis Identified Hormone-Related Genes in Dormancy Release
3.10. RT-qPCR Verification
4. Discussion
5. Perspective
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lang, G.A.; Early, J.D.; Martin, G.C.; Darnell, R.L. Endo-, Para-, and Ecodormancy: Physiological Terminology and Classification for Dormancy Research. Hortscience 1987, 22, 271–277. [Google Scholar]
- Cline, M.G.; Deppong, D.O. The Role of Apical Dominance in Paradormancy of Temperate Woody Plants: A Reappraisal. J. Plant Physiol. 1999, 155, 350–356. [Google Scholar] [CrossRef]
- Qin, Z.; Bai, Y.; Muhammad, S.; Wu, X.; Deng, P.; Wu, J.; An, H.; Wu, L. Divergent roles of FT-like 9 in flowering transition under different day lengths in Brachypodium distachyon. Nat. Commun. 2019, 10, 812. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, W.; Gao, Z.; Wang, L.; Zhong, W.; Ni, Z.; Zhang, Z. Comparative proteomic and transcriptomic approaches to address the active role of GA4 in Japanese apricot flower bud dormancy release. J. Exp. Bot. 2013, 64, 4953–4966. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, N.; Liu, R.; Chen, M.; Zhang, J. H2O2 mediates the regulation of ABA catabolism and GA biosynthesis in Arabidopsis seed dormancy and germination. J. Exp. Bot. 2010, 61, 2979–2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Li, W.; Zheng, P.; Xu, T.; Chen, L.; Liu, D.; Hussain, S.; Teng, Y. Transcriptomic analysis of ‘Suli’ pear (Pyrus pyrifolia white pear group) buds during the dormancy by RNA-Seq. BMC Genom. 2012, 13, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Yang, Q.; Yan, X.; Wu, X.; Yang, F.; Li, J.; Wei, J.; Ni, J.; Ahmad, M.; Bai, S.; et al. High-quality genome assembly of ‘Cuiguan’ pear (Pyrus pyrifolia) as a reference genome for identifying regulatory genes and epigenetic modifications responsible for bud dormancy. Hortic. Res. 2021, 8, 197. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Xin, D.; Chen, W.; Shao, X.; Wang, Y.; Guo, W. RNA-Seq-based transcriptome analysis of dormant flower buds of Chinese cherry (Prunus pseudocerasus). Gene 2015, 555, 362–376. [Google Scholar] [CrossRef]
- Ionescu, I.A.; López-Ortega, G.; Burow, M.; Bayo-Canha, A.; Junge, A.; Gericke, O.; Møller, B.L.; Sánchez-Pérez, R. Transcriptome and Metabolite Changes during Hydrogen Cyanamide-Induced Floral Bud Break in Sweet Cherry. Front. Plant Sci. 2017, 8, 1233. [Google Scholar] [CrossRef]
- Yu, J.; Conrad, A.O.; Decroocq, V.; Zhebentyayeva, T.; Williams, D.E.; Bennett, D.; Roch, G.; Audergon, J.-M.; Dardick, C.; Liu, Z.; et al. Distinctive Gene Expression Patterns Define Endodormancy to Ecodormancy Transition in Apricot and Peach. Front. Plant Sci. 2020, 11, 180. [Google Scholar] [CrossRef] [Green Version]
- Khalil-Ur-Rehman, M.; Sun, L.; Li, C.-X.; Faheem, M.; Wang, W.; Tao, J.-M. Comparative RNA-seq based transcriptomic analysis of bud dormancy in grape. BMC Plant Biol. 2017, 17, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, Z.; Zhao, X.; Li, R.; Yang, B.; Liu, M.; Fang, Y. Comparative transcriptome analysis provides insight into differentially expressed genes related to bud dormancy in grapevine (Vitis vinifera). Sci. Hortic. 2017, 225, 213–220. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Lai, B.; Xia, H.; Wang, H.; Huang, X. Morphological Characterization and Gene Expression Profiling during Bud Development in a Tropical Perennial, Litchi chinensis Sonn. Front. Plant Sci. 2016, 7, 1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Q.; Li, J.; Cai, D.; Qian, M.; Jia, H.; Bai, S.; Hussain, S.; Liu, G.; Teng, Y.; Zheng, X. Dormancy-associated MADS-box genes and microRNAs jointly control dormancy transition in pear (Pyrus pyrifoliawhite pear group) flower bud. J. Exp. Bot. 2016, 67, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Bai, S.; Imai, T.; Ito, A.; Nakajima, I.; Moriguchi, T. Histone modification and signalling cascade of thedormancy-associated MADS-boxgene, PpMADS13-1, in Japanese pear (Pyrus pyrifolia) during endodormancy. Plant Cell Environ. 2014, 38, 1157–1166. [Google Scholar] [CrossRef]
- Yang, Q.; Yang, B.; Li, J.; Wang, Y.; Tao, R.; Yang, F.; Wu, X.; Yan, X.; Ahmad, M.; Shen, J.; et al. ABA-responsive ABRE-BINDING FACTOR3 activates DAM3 expression to promote bud dormancy in Asian pear. Plant Cell Environ. 2020, 43, 1360–1375. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, G.; Zhao, X.; Zhang, Z.; Wang, X.; Liu, X.; Xiao, W.; Fu, X.; Chen, X.; Gao, D.; et al. Transcription factor TCP20 regulates peach bud endodormancy by inhibiting DAM5/DAM6 and interacting with ABF2. J. Exp. Bot. 2019, 71, 1585–1597. [Google Scholar] [CrossRef]
- Singh, R.; Maurya, J.P.; Azeez, A.; Miskolczi, P.; Tylewicz, S.; Stojkovič, K.; Delhomme, N.; Busov, V.; Bhalerao, R.P. A genetic network mediating the control of bud break in hybrid aspen. Nat. Commun. 2018, 9, 4173. [Google Scholar] [CrossRef] [Green Version]
- Lobos, G.A.; Hancock, J.F. Breeding blueberries for a changing global environment: A review. Front. Plant Sci. 2015, 6, 782. [Google Scholar] [CrossRef] [Green Version]
- Bigler, C.; Bugmann, H. Climate-induced shifts in leaf unfolding and frost risk of European trees and shrubs. Sci. Rep. 2018, 8, 9865. [Google Scholar] [CrossRef] [Green Version]
- Legave, J.-M.; Guédon, Y.; Malagi, G.; EL Yaacoubi, A.; Bonhomme, M. Differentiated Responses of Apple Tree Floral Phenology to Global Warming in Contrasting Climatic Regions. Front. Plant Sci. 2015, 6, 1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, X.; Yang, Y.; Yue, C.; Wang, L.; Horvath, D.P.; Wang, X. Comprehensive Transcriptome Analyses Reveal Differential Gene Expression Profiles of Camellia sinensis Axillary Buds at Para-, Endo-, Ecodormancy, and Bud Flush Stages. Front. Plant Sci. 2017, 8, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, E.A. A Model for Estimating the Completion of Rest for “Redhaven” and “Elberta” Peach Trees. HortScience 1974, 9, 331–332. [Google Scholar]
- Yooyongwech, S.; Sugaya, S.; Sekozawa, Y.; Gemma, H. Differential adaptation of high- and low-chill dormant peaches in winter through aquaporin gene expression and soluble sugar content. Plant Cell Rep. 2009, 28, 1709–1715. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Tarazona, S.; García, F.; Ferrer, A.; Dopazo, J.; Conesa, A. NOIseq: A RNA-seq differential expression method robust for sequencing depth biases. EMBnet. J. 2012, 17, 18. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, G.; Götz, S.; Conesa, A.; Segrelles, D.; Blanquer, I.; García, J.M.; Hernandez, V.; Robles, M.; Talon, M. Blast2GO goes grid: Developing a grid-enabled prototype for functional genomics analysis. Stud. Health Technol. Inf. 2006, 2006, 120. [Google Scholar]
- Minoru, K.; Michihiro, A.; Susumu, G.; Masahiro, H.; Mika, H.; Masumi, I.; Toshiaki, K.; Shuichi, K.; Shujiro, O.; Toshiaki, T. KEGG for Linking Genomes to Life and the Environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Zhaoyun, C.; Youning, M.; Renxiang, M.; Shasha, Y.; Mingxue, C. Analysis of 17 Cytokinins in Rice by Solid Phase Extraction Purification and Liquid Chromatography-Tandem Mass Spectrometry. Chin. J. Chromatogr. 2015, 33, 715–721. [Google Scholar]
- Zhao, J.; Yu, N.; Ju, M.; Fan, B.; Zhang, Y.; Zhu, E.; Zhang, M.; Zhang, K. ABC transporter OsABCG18 controls the shootward transport of cytokinins and grain yield in rice. J. Exp. Bot. 2019, 70, 6277–6291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šimura, J.; Antoniadi, I.; Široká, J.; Tarkowská, D.; Strnad, M.; Ljung, K.; Novák, O. Plant Hormonomics: Multiple Phytohormone Profiling by Targeted Metabolomics. Plant Physiol. 2018, 177, 476–489. [Google Scholar] [CrossRef] [Green Version]
- Hellens, R.P.; Allan, A.C.; Friel, E.N.; Bolitho, K.; Grafton, K.; Templeton, M.D.; Karunairetnam, S.; Gleave, A.P.; Laing, W.A. Transient expression vectors for functional genomics, quantification of promoter activity and RNA silencing in plants. Plant Methods 2005, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Chen, M.; Cai, K.; Zhang, L.; Zhu, Y.; Ye, Q.; Lü, M.; Liao, F.; Chen, W.; Guo, W. VcFAS, VcSUN and VcOVATE orchestrated the fruit morphogenesis in southern highbush blueberry during the pre-anthesis and fruit development. Sci. Hortic. 2018, 240, 109–115. [Google Scholar] [CrossRef]
- Die, J.V.; Rowland, L.J. Superior Cross-Species Reference Genes: A Blueberry Case Study. PLoS ONE 2013, 8, e73354. [Google Scholar] [CrossRef]
- Yang, L.; Liu, L.; Wang, Z.; Zong, Y.; Yu, L.; Li, Y.; Liao, F.; Chen, M.; Cai, K.; Guo, W. Comparative anatomical and transcriptomic insights into Vaccinium corymbosum flower bud and fruit throughout development. BMC Plant Biol. 2021, 21, 289. [Google Scholar] [CrossRef]
- Bai, S.; Saito, T.; Sakamoto, D.; Ito, A.; Fujii, H.; Moriguchi, T. Transcriptome Analysis of Japanese Pear (Pyrus pyrifolia Nakai) Flower Buds Transitioning Through Endodormancy. Plant Cell Physiol. 2013, 54, 1132–1151. [Google Scholar] [CrossRef] [Green Version]
- Ruttink, T.; Arend, M.; Morreel, K.; Storme, V.; Rombauts, S.; Fromm, J.; Bhalerao, R.; Boerjan, W.; Rohde, A. A Molecular Timetable for Apical Bud Formation and Dormancy Induction in Poplar. Plant Cell 2007, 19, 2370–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangguan, L.; Chen, M.; Fang, X.; Xie, Z.; Gong, P.; Huang, Y.; Wang, Z.; Fang, J. Comparative transcriptome analysis provides insight into regulation pathways and temporal and spatial expression characteristics of grapevine (Vitis vinifera) dormant buds in different nodes. BMC Plant Biol. 2020, 20, 390. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Martínez, J.A.; Poza-Carrión, C.; Cubas, P. Arabidopsis BRANCHED1Acts as an Integrator of Branching Signals within Axillary Buds. Plant Cell 2007, 19, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.E.K.; Eriksson, M.E.; Junttila, O. The dynamic nature of bud dormancy in trees: Environmental control and molecular mechanisms. Plant Cell Environ. 2012, 35, 1707–1728. [Google Scholar] [CrossRef] [PubMed]
- Pin, P.A.; Nilsson, O. The multifaceted roles of FLOWERING LOCUS T in plant development. Plant Cell Environ. 2012, 35, 1742–1755. [Google Scholar] [CrossRef]
- Böhlenius, H.; Huang, T.; Charbonnel-Campaa, L.; Brunner, A.M.; Jansson, S.; Strauss, S.H.; Nilsson, O. CO/FT Regulatory Module Controls Timing of Flowering and Seasonal Growth Cessation in Trees. Science 2006, 312, 1040–1043. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Li, T.; Xu, P.-B.; Li, L.; Du, S.-S.; Lian, H.-L.; Yang, H.-Q. DELLA proteins physically interact with CONSTANS to regulate flowering under long days inArabidopsis. FEBS Lett. 2016, 590, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Zhongyuan, B.; Yu, Y.; Zepeng, L.; Yanchao, L.; Wen, J.; Ying, H.; Ai-Wu, D. Regulation of Arabidopsis Flowering by the Histone Mark Readers MRG1/2 via Interaction with CONSTANS to Modulate FT Expression. PLoS Genet. 2014, 10, e1004617. [Google Scholar]
- Ben Mohamed, H.; Vadel, A.M.; Geuns, J.M.; Khemira, H. Biochemical changes in dormant grapevine shoot tissues in response to chilling: Possible role in dormancy release. Sci. Hortic. 2010, 124, 440–447. [Google Scholar] [CrossRef]
- Ben Mohamed, H.; Vadel, A.M.; Geuns, J.M.; Khemira, H. Carbohydrate changes during dormancy release in Superior Seedless grapevine cuttings following hydrogen cyanamide treatment. Sci. Hortic. 2012, 140, 19–25. [Google Scholar] [CrossRef]
- Guak, S.; Fuchigami, L.H. Effects of Applied ABA on Growth Cessation, Bud Dormancy, Cold Acclimation, Leaf Senescence and N Mobilization in Apple Nursery Plants. J. Hortic. Sci. Biotechnol. 2001, 76, 459–464. [Google Scholar] [CrossRef]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C Phosphatase Activity Function as Abscisic Acid Sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, Y.; Niu, Q.; He, L.; Teng, Y.; Bai, S. Abscisic Acid (ABA ) Promotes the Induction and Maintenance of Pear (Pyrus pyrifolia White Pear Group) Flower Bud Endodormancy. Int. J. Mol. Sci. 2018, 19, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnusamy, V.; Zhu, J.; Zhu, J.-K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Puig, C.P.; Dagar, A.; Ibanez, C.M.; Singh, V.; Crisosto, C.H.; Friedman, H.; Lurie, S.; Granell, A. Pre-symptomatic transcriptome changes during cold storage of chilling sensitive and resistant peach cultivars to elucidate chilling injury mechanisms. BMC Genom. 2015, 16, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, M.; Zhou, B.; Yang, Y.; Wei, Q.; Zhang, J. Transcriptome analysis provides insights into the stress response crosstalk in apple (Malus × domestica) subjected to drought, cold and high salinity. Sci. Rep. 2019, 9, 9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, S.; GAO, Y.; Maidunur, Y.; PAN, Y.; SHAO, W.; ZONG, Y.; CHEN, W.; YANG, L.; GUO, W.; LI, Y. Research on Application Exogenous Abscisic Acid in Inhibiting Early Flowering and Associated Genes Expression Characteristics in Blueberry. J. Fruit Sci. 2020, 38, 325–334. [Google Scholar] [CrossRef]
- Davies, P.J. The Plant Hormones: Their Nature, Occurrence, and Functions. In Plant Hormones; Springer: Dordrecht, The Netherlands, 1995. [Google Scholar]
- Howe, G.T.; Horvath, D.P.; Dharmawardhana, P.; Priest, H.D.; Mockler, T.C.; Strauss, S.H. Extensive Transcriptome Changes During Natural Onset and Release of Vegetative Bud Dormancy in Populus. Front. Plant Sci. 2015, 6, 989. [Google Scholar] [CrossRef] [Green Version]
- Bleecker, A.B.; Kende, H. Ethylene: A Gaseous Signal Molecule in Plants. Annu. Rev. Cell Dev. Biol. 2000, 16, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Stepanova, A.N.; Alonso, J.M. Alonso Ethylene Signalling and Response Pathway: A Unique Signalling Cascade with a Multitude of Inputs and Outputs. Physiol. Plant. 2005, 123, 195–206. [Google Scholar] [CrossRef]
- Arenas, S.; Cortés, A.J.; Mastretta-Yanes, A.; Jaramillo-Correa, J.P. Evaluating the accuracy of genomic prediction for the management and conservation of relictual natural tree populations. Tree Genet. Genomes 2021, 17, 12. [Google Scholar] [CrossRef]
- Cortés, A.J.; Restrepo-Montoya, M.; Bedoya-Canas, L.E. Modern Strategies to Assess and Breed Forest Tree Adaptation to Changing Climate. Front. Plant Sci. 2020, 11, 583323. [Google Scholar] [CrossRef] [PubMed]
- Samish, M.R. Dormancy in Woody Plants. Annu. Rev. Plant Physiol. 1954, 5, 183–204. [Google Scholar] [CrossRef]
- Nishiyama, S.; Fujikawa, M.; Yamane, H.; Shirasawa, K.; Babiker, E.; Tao, R. Genomic insight into the developmental history of southern highbush blueberry populations. Heredity 2020, 126, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Medina Cano, C.I.; Lobo Arias, M.; Castaño Colorado, Á.A.; Cardona, L.E. Development analysis of Mortiño (Vaccinium meridionale Swart.) plants derived from clonal and sexual propagation. CTA 2015, 16, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Cappai, F.; Garcia, A.; Cullen, R.; Davis, M.; Munoz, P.R. Advancements in Low-Chill Blueberry Vaccinium corymbosum L. Tissue Culture Practices. Plants 2020, 9, 1624. [Google Scholar] [CrossRef]
- Frías-Ortega, C.E.; Alejo-Santiago, G.; Bugarín-Montoya, R.; Aburto-González, C.A.; Juárez-Rosete, C.R.; Urbina-Sánchez, E.; Sánchez-Hernández, E. Nutrient Solution Concentration and Its Relationship with Blueberry Production and Quality. Cienc. Tecnol. Agropecu. 2020, 21, e1296. [Google Scholar]
- Yang, Q.; Gao, Y.; Wu, X.; Moriguchi, T.; Bai, S.; Teng, Y. Bud endodormancy in deciduous fruit trees: Advances and prospects. Hortic. Res. 2021, 8, 139. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Data | Clean Data | Clean Data Ratio (%) | Clean Data Q20 (%) | Clean Data Q30 (%) | GC Content (%) | Mapped Reads | Percentage of Mapped Reads | Mapped Genome (%) | Mapped Gene (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Nov. 19-1 | 41,082,384 | 41,078,456 | 99.99 | 97.53 | 93.91 | 46.84 | 29,594,336 | 72.04% | 72.04% | 59.60% |
| Nov. 19-2 | 40,638,858 | 40,635,402 | 99.99 | 97.48 | 93.82 | 47.12 | 32,048,468 | 78.87% | 78.87% | 68.31% |
| Nov. 19-3 | 41,003,184 | 40,999,156 | 99.99 | 97.47 | 93.81 | 47.3 | 32,364,244 | 78.94% | 78.94% | 68.54% |
| Dec. 8-1 | 40,359,734 | 40,354,476 | 99.99 | 97.36 | 93.63 | 47.8 | 29,287,920 | 72.58% | 72.58% | 61.09% |
| Dec. 8-2 | 40,379,528 | 40,373,466 | 99.98 | 97.27 | 93.47 | 47.71 | 30,525,004 | 75.61% | 75.61% | 63.26% |
| Dec. 8-3 | 40,117,154 | 40,111,158 | 99.99 | 97.57 | 94.06 | 47.59 | 30,338,448 | 75.64% | 75.64% | 62.35% |
| Dec. 29-1 | 41,356,720 | 41,351,040 | 99.99 | 97.67 | 94.28 | 47.28 | 32,701,506 | 79.08% | 79.08% | 65.14% |
| Dec. 29-2 | 41,127,706 | 41,122,630 | 99.99 | 97.62 | 94.13 | 47.28 | 32,066,614 | 77.98% | 77.98% | 63.66% |
| Dec. 29-3 | 41,169,840 | 41,163,932 | 99.99 | 97.61 | 94.11 | 47.64 | 32,503,736 | 78.96% | 78.96% | 66.26% |
| 6 h-1 | 40,431,514 | 40,428,068 | 99.99 | 97.83 | 94.5 | 46.61 | 29,676,104 | 73.40% | 73.40% | 59.28% |
| 6 h-2 | 40,489,884 | 40,486,186 | 99.99 | 97.23 | 93.16 | 46.69 | 30,991,372 | 76.55% | 76.62% | 65.72% |
| 6 h-3 | 40,486,658 | 40,482,478 | 99.99 | 97.19 | 93.08 | 47.3 | 31,017,546 | 76.62% | 76.55% | 64.33% |
| 12 h-1 | 41,321,014 | 41,316,892 | 99.99 | 97.31 | 93.31 | 47.08 | 32,476,742 | 78.60% | 78.60% | 68.04% |
| 12 h-2 | 40,172,084 | 40,168,200 | 99.99 | 97.3 | 93.31 | 47.24 | 30,641,344 | 76.28% | 76.28% | 65.73% |
| 12 h-3 | 41,155,516 | 41,149,572 | 99.99 | 97.2 | 93.16 | 47.77 | 32,798,908 | 79.71% | 79.71% | 68.83% |
| 18 h-1 | 40,407,682 | 40,404,328 | 99.99 | 97.41 | 93.53 | 46.63 | 29,553,020 | 73.14% | 73.14% | 61.94% |
| 18 h-2 | 40,400,518 | 40,397,044 | 99.99 | 97.31 | 93.3 | 46.66 | 29,263,828 | 72.44% | 72.44% | 61.06% |
| Gene ID | GO | KEGG Annotation |
|---|---|---|
| VaccDscaff33-processed-gene-241.9 | GO:0050794 regulation of cellular process | ethylene response factor 12 |
| VaccDscaff43-augustus-gene-228.38 | GO:0045893 positive regulation of transcription | elongation factor G-2 |
| VaccDscaff8-snap-gene-394.21 | GO:0007165 signal transduction|GO:0006351 transcription | two-component response regulator ARR1 isoform X2 |
| VaccDscaff13-augustus-gene-302.19 | GO:0042787 protein ubiquitination involved in ubiquitin-dependent protein catabolic process | E3 ubiquitin-protein ligase UPL3 |
| VaccDscaff46-augustus-gene-87.38 | GO:0008152 metabolic process | ABC transporter F family member 1 isoform X1 |
| VaccDscaff46-augustus-gene-19.24 | GO:0045893 positive regulation of transcription | elongation factor G-2, chloroplastic |
| VaccDscaff47-augustus-gene-75.30 | GO:0048519 negative regulation of biological process | homeobox-leucine zipper protein REVOLUTA |
| VaccDscaff5-augustus-gene-239.20 | ----- | E3 ubiquitin protein ligase RIE1-like |
| VaccDscaff703-augustus-gene-0.9 | GO:0006355 regulation of transcription, DNA-templated | homeobox-leucine zipper protein HOX11-like |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Ma, R.; Li, R.; Zhao, Q.; Zhang, Z.; Zong, Y.; Yao, L.; Chen, W.; Yang, L.; Liao, F.; et al. Comparative Transcriptomic Analysis Provides Insight into the Key Regulatory Pathways and Differentially Expressed Genes in Blueberry Flower Bud Endo- and Ecodormancy Release. Horticulturae 2022, 8, 176. https://doi.org/10.3390/horticulturae8020176
Li Y, Ma R, Li R, Zhao Q, Zhang Z, Zong Y, Yao L, Chen W, Yang L, Liao F, et al. Comparative Transcriptomic Analysis Provides Insight into the Key Regulatory Pathways and Differentially Expressed Genes in Blueberry Flower Bud Endo- and Ecodormancy Release. Horticulturae. 2022; 8(2):176. https://doi.org/10.3390/horticulturae8020176
Chicago/Turabian StyleLi, Yongqiang, Rui Ma, Ruixue Li, Qi Zhao, Zhenzhen Zhang, Yu Zong, Linbo Yao, Wenrong Chen, Li Yang, Fanglei Liao, and et al. 2022. "Comparative Transcriptomic Analysis Provides Insight into the Key Regulatory Pathways and Differentially Expressed Genes in Blueberry Flower Bud Endo- and Ecodormancy Release" Horticulturae 8, no. 2: 176. https://doi.org/10.3390/horticulturae8020176
APA StyleLi, Y., Ma, R., Li, R., Zhao, Q., Zhang, Z., Zong, Y., Yao, L., Chen, W., Yang, L., Liao, F., Zhu, Y., & Guo, W. (2022). Comparative Transcriptomic Analysis Provides Insight into the Key Regulatory Pathways and Differentially Expressed Genes in Blueberry Flower Bud Endo- and Ecodormancy Release. Horticulturae, 8(2), 176. https://doi.org/10.3390/horticulturae8020176