
Anthocyanin Metabolite and Transcriptome Sequencing Analysis Reveals White Flowers in Rhododendron dauricum var. albiflorum
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Extraction, Identification, and Quantification of Anthocyanin Metabolites
2.3. Transcriptome Sequencing
2.4. Structure and Functional Annotation of Transcripts
2.5. Differentially Expressed Gene (DEG) Analysis
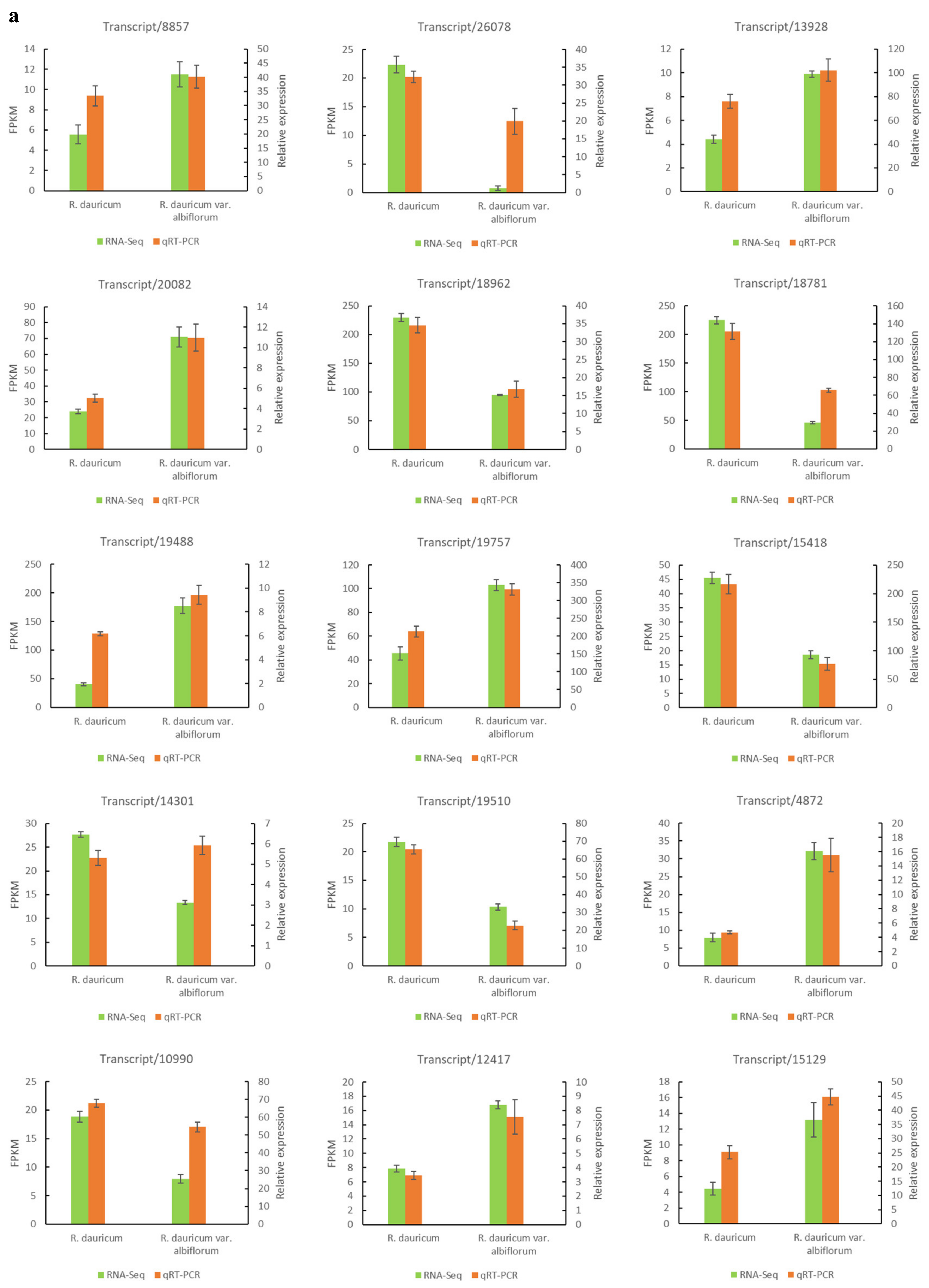
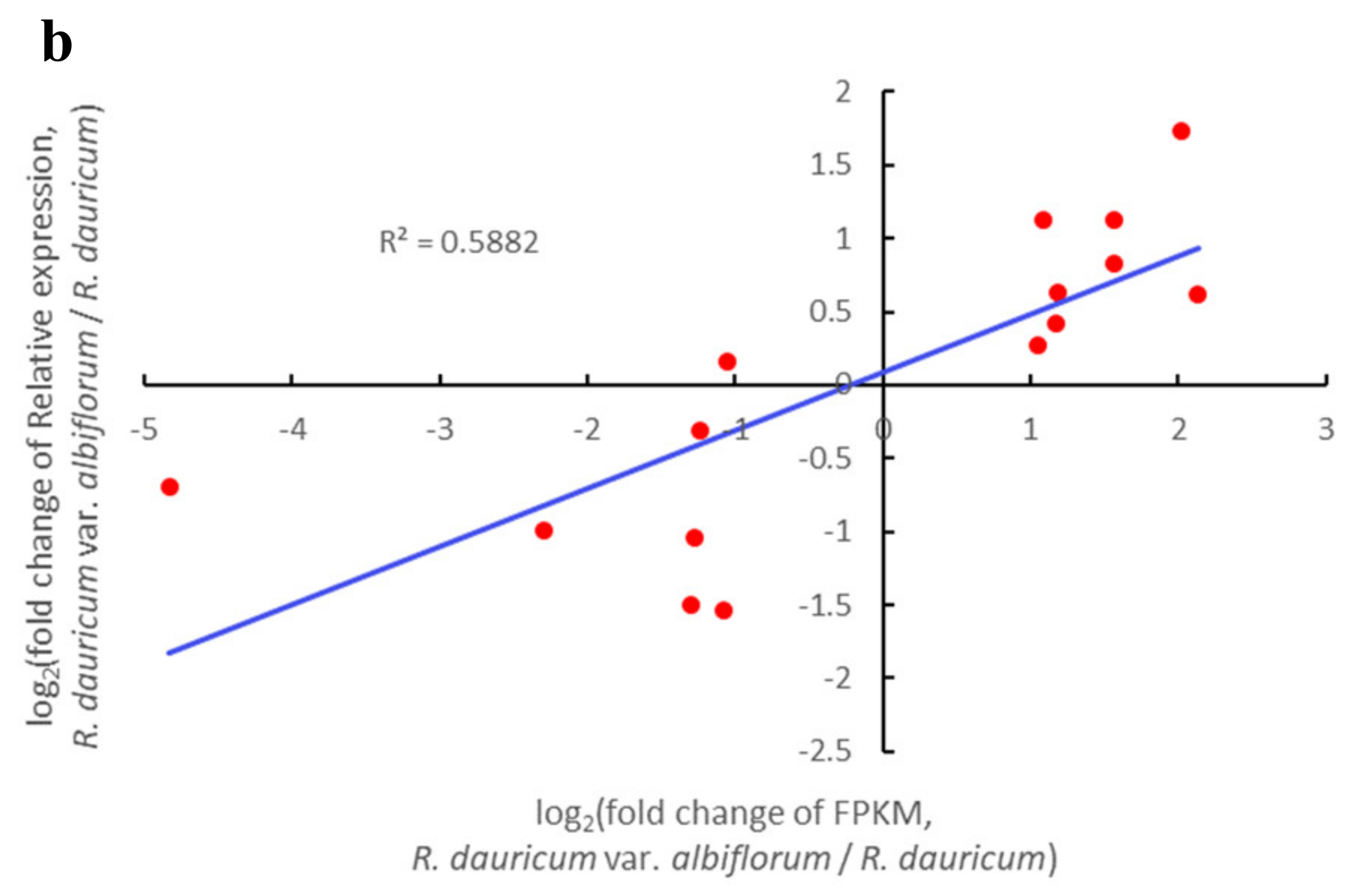
2.6. Quantitative Real-Time PCR (qRT-PCR) Validation
3. Results
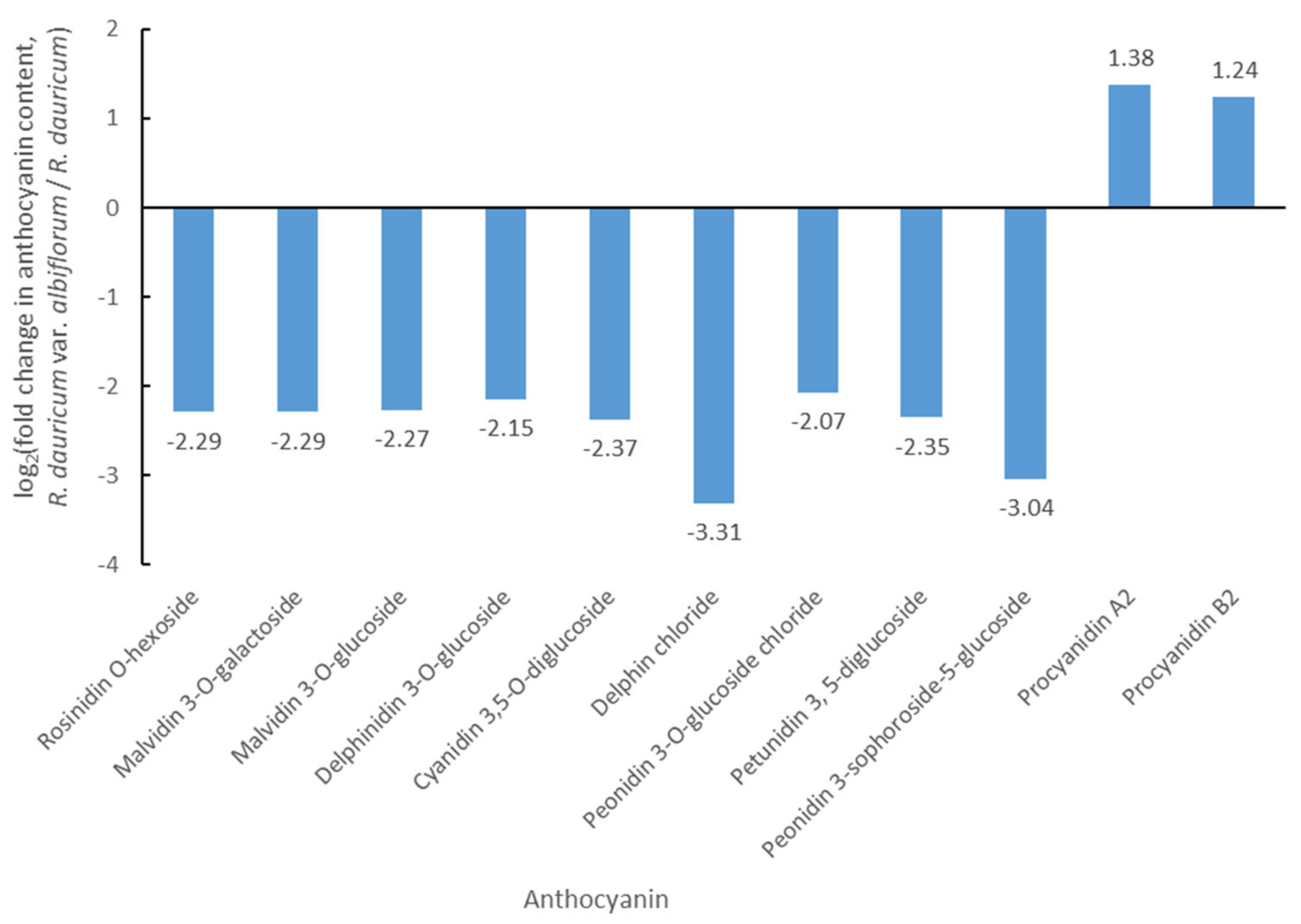
3.1. Differential Accumulation of Anthocyanins
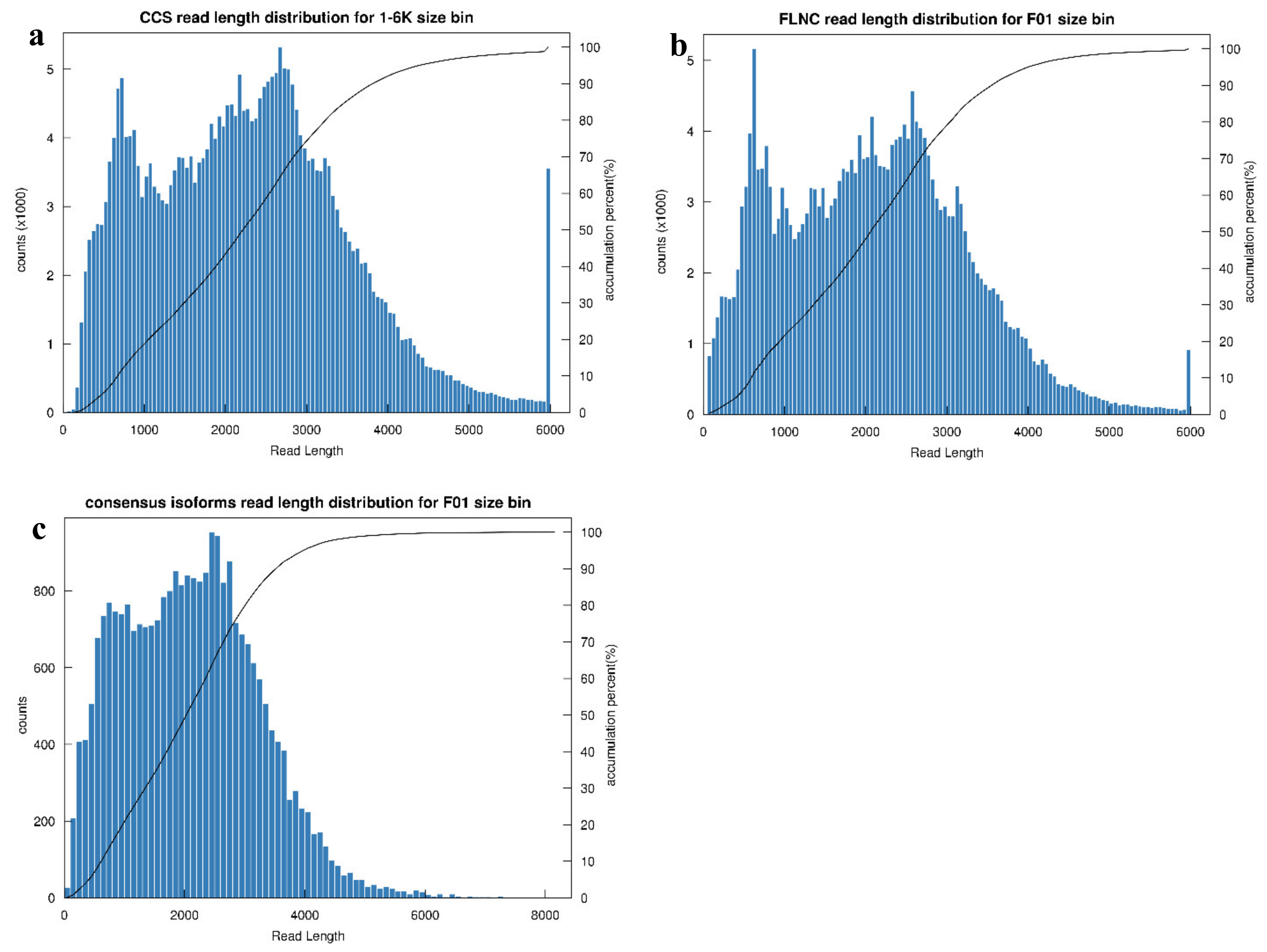
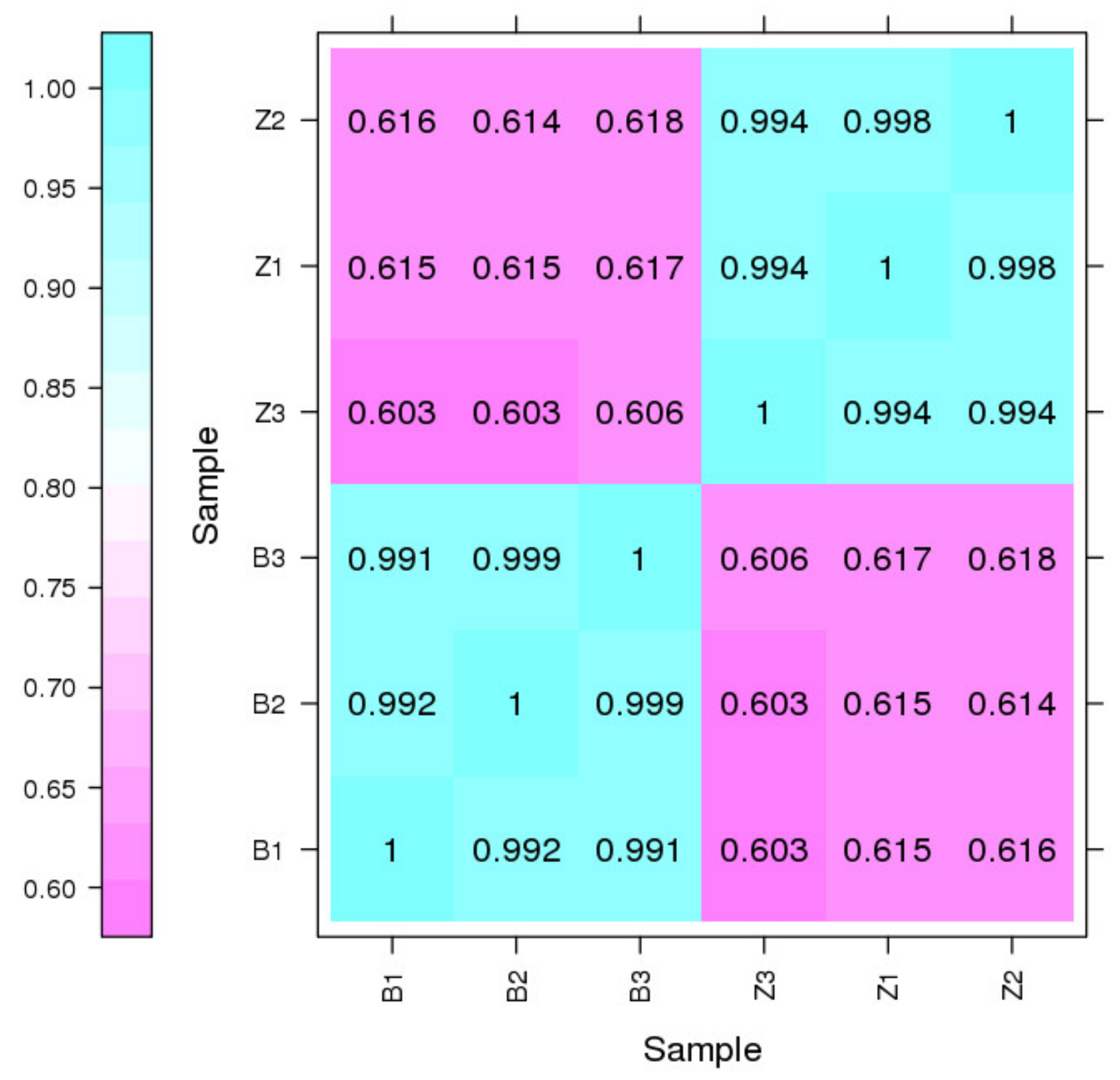
3.2. General Properties of Transcriptome Sequencing
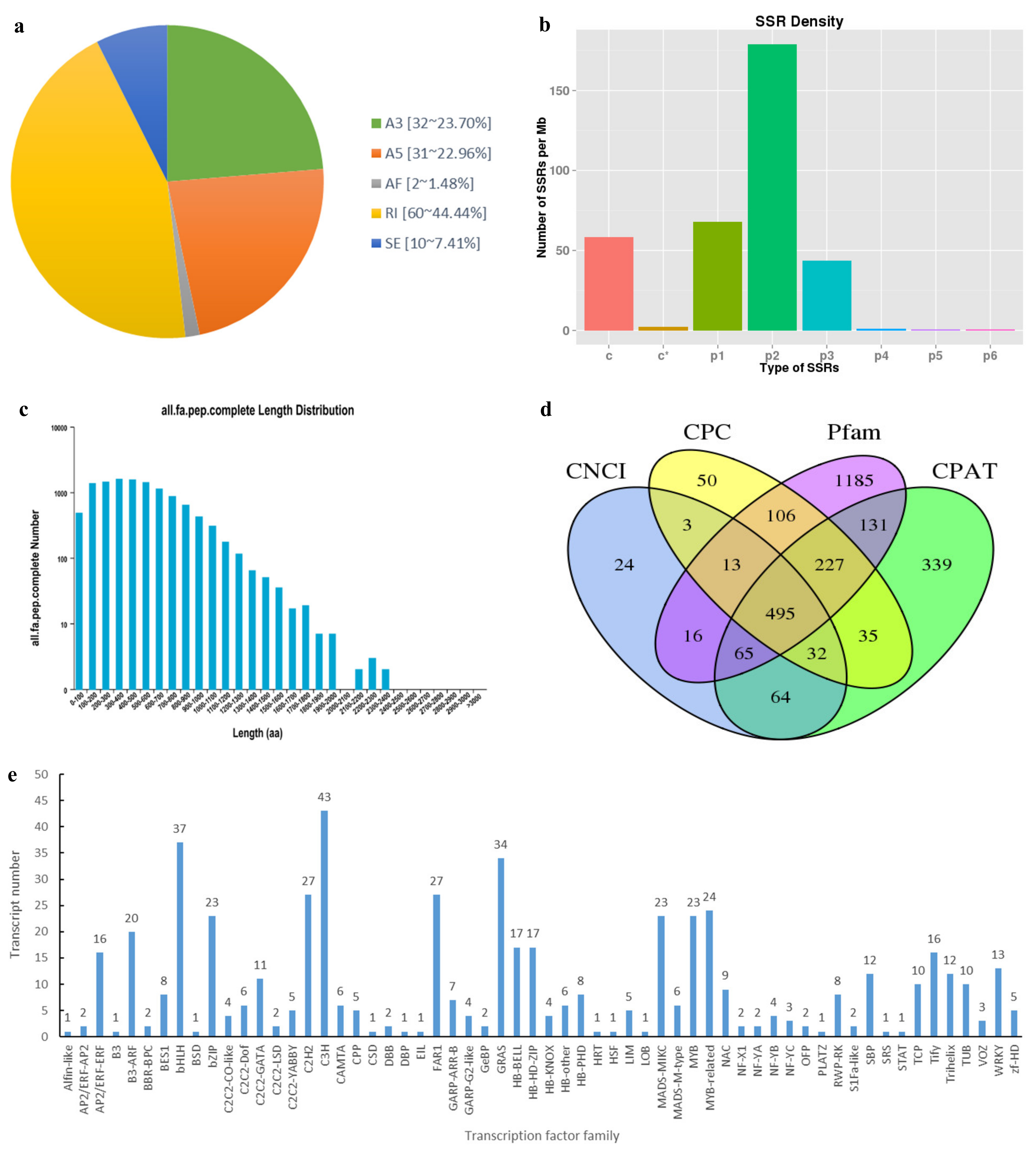
3.3. Characterization of Transcript Structure
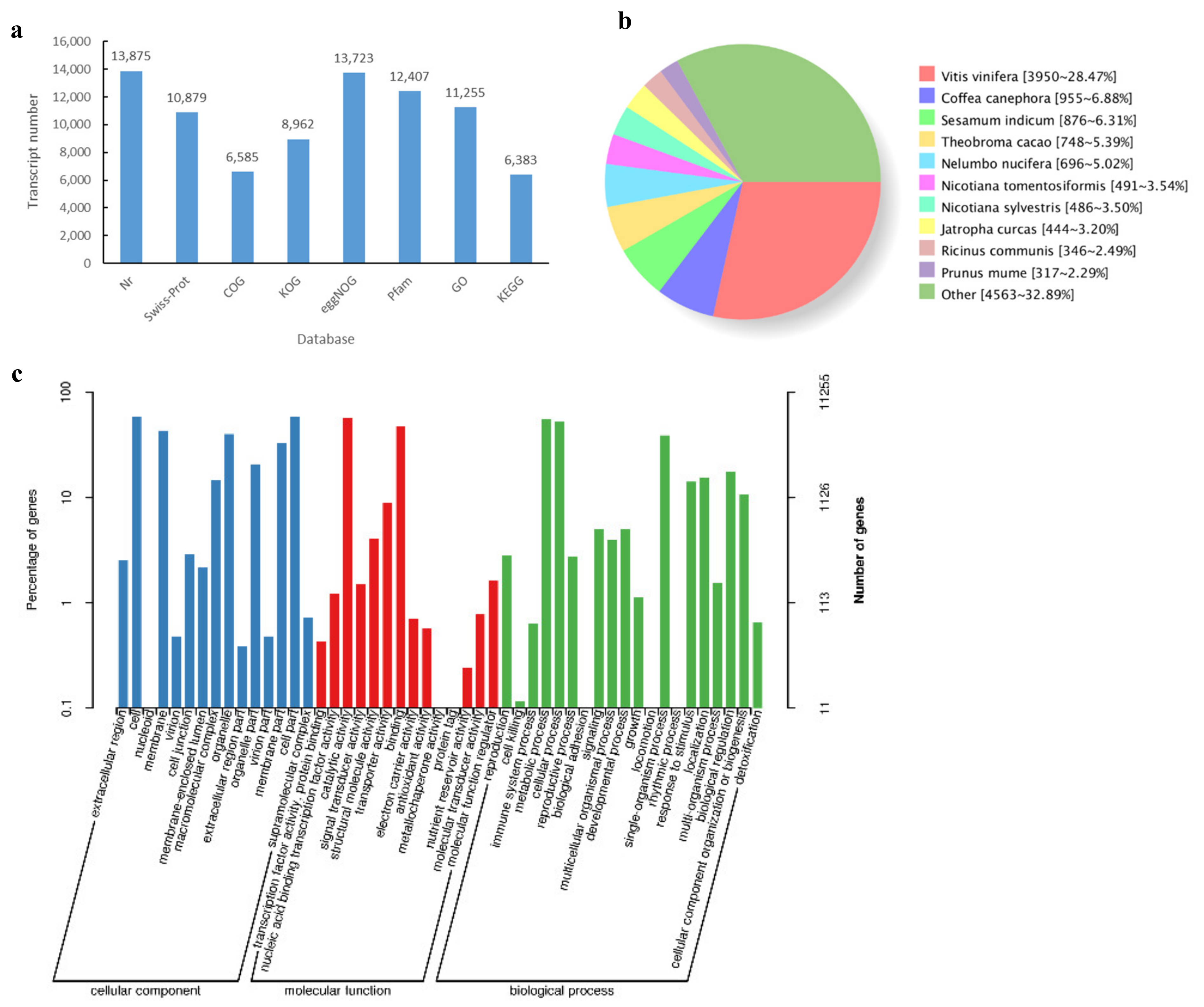
3.4. Functional Annotation of Transcripts
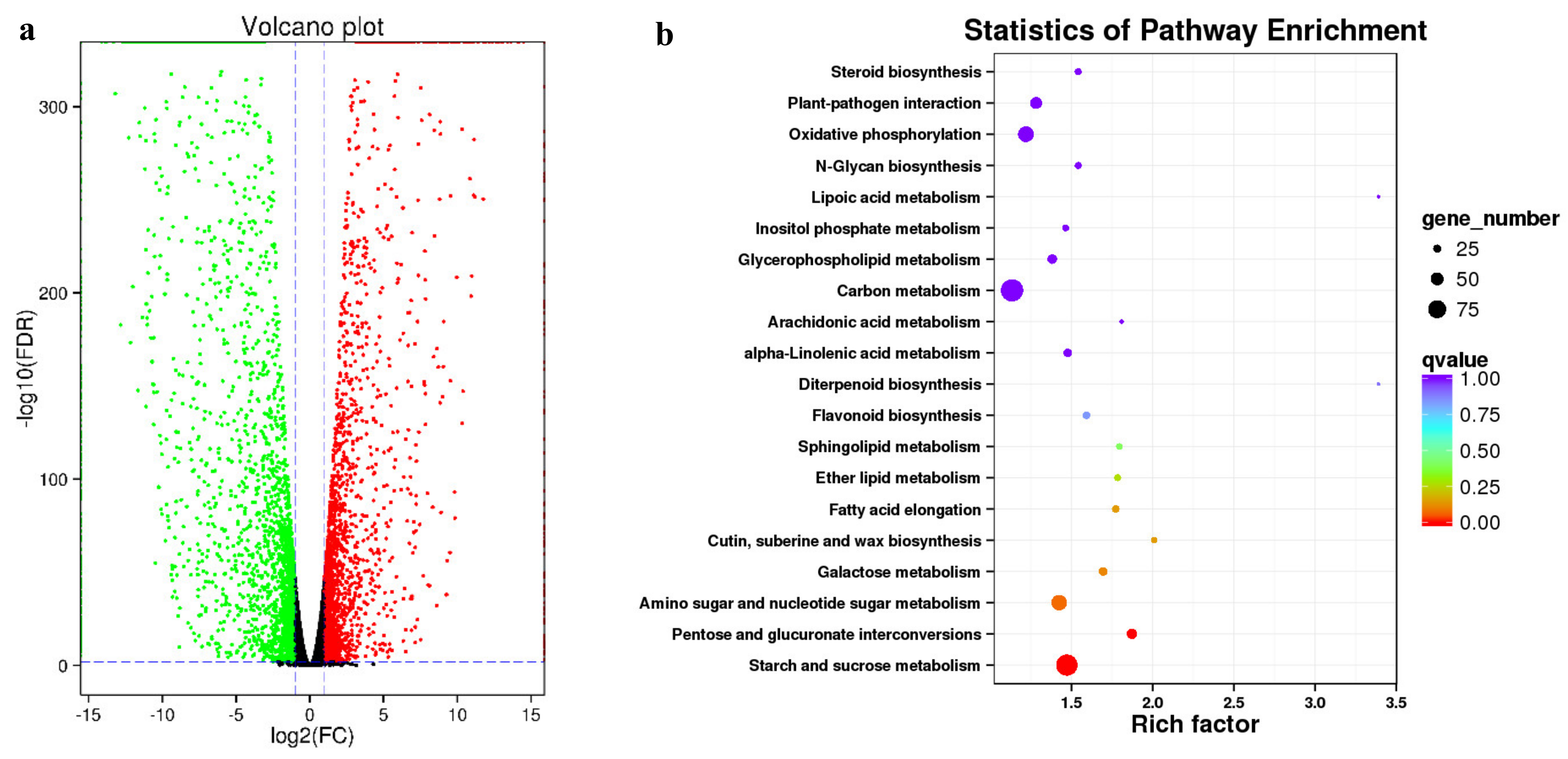
3.5. Identification and Enrichment of DEGs
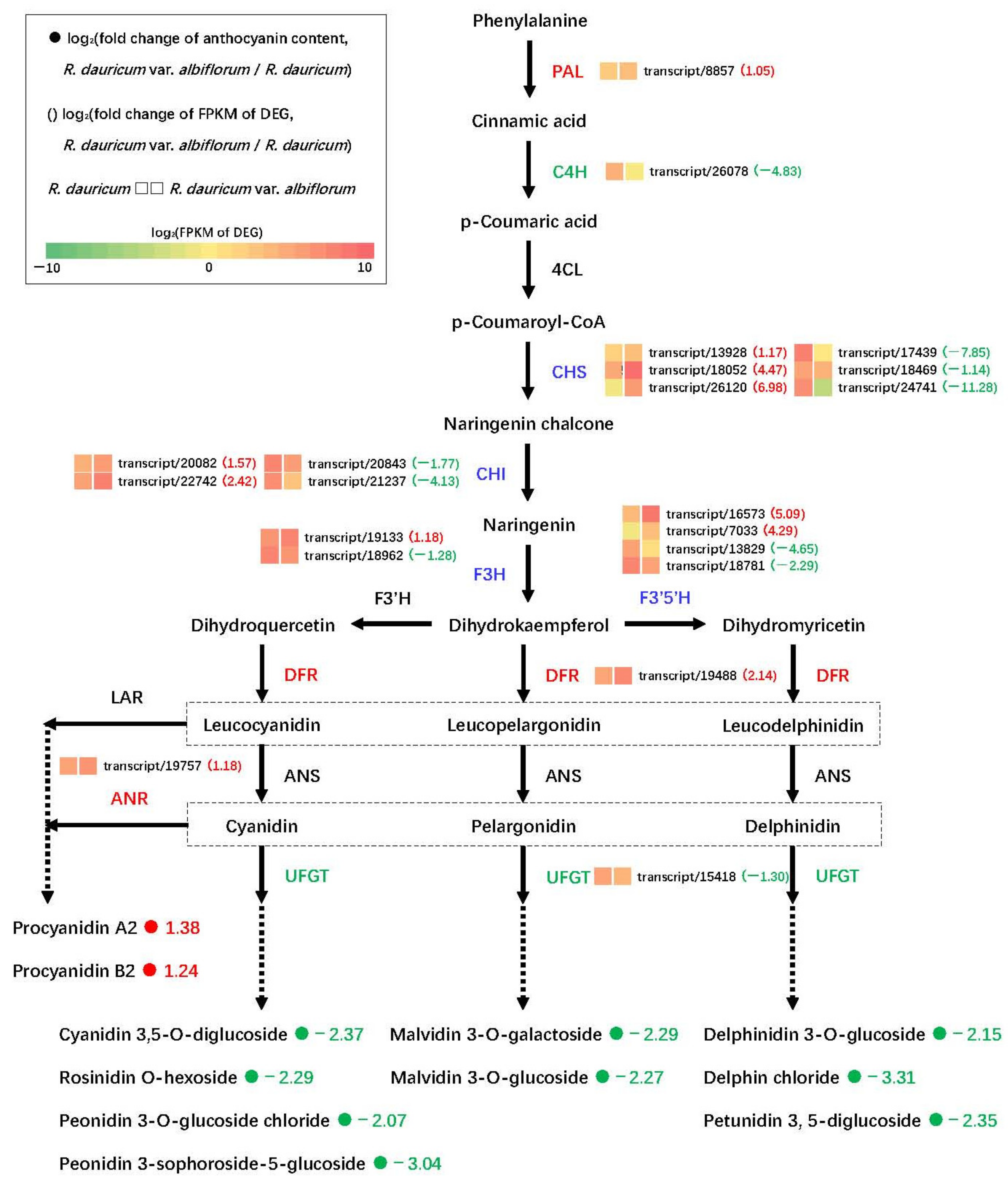
3.6. DEGs Related to Flower Color
4. Discussion
4.1. Anthocyanin Identification in R. dauricum var. albiflorum
4.2. Anthocyanin Biosynthesis Responsible for White Flower of R. dauricum var. albiflorum
4.3. TFs Related to Anthocyanin Biosynthesis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kishi-Kaboshi, M.; Aida, R.; Sasaki, K. Genome engineering in ornamental plants: Current status and future prospects. Plant Physiol. Biochem. 2018, 131, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Mekapogu, M.; Vasamsetti, B.M.K.; Kwon, O.K.; Ahn, M.S.; Lim, S.H.; Jung, J.A. Anthocyanins in floral colors: Biosynthesis and regulation in Chrysanthemum flowers. Int. J. Mol. Sci. 2020, 21, 6537. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Jiao, F.C.; Zhao, L.; Wu, X.F.; Song, Z.B.; Li, Y.P. Metabolome and transcriptome analyses of the molecular mechanisms of flower color mutation in tobacco. BMC Genom. 2020, 21, 611. [Google Scholar] [CrossRef]
- Liu, W.X.; Feng, Y.; Yu, S.H.; Fan, Z.Q.; Li, X.L.; Li, J.Y.; Yin, H.F. The flavonoid biosynthesis network in plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef]
- Petroni, K.; Tonelli, C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011, 181, 219–229. [Google Scholar] [CrossRef]
- LaFountain, A.M.; Yuan, Y.W. Repressors of anthocyanin biosynthesis. New Phytol. 2021, 231, 933–949. [Google Scholar] [CrossRef]
- Saigo, T.; Wang, T.; Watanabe, M.; Tohge, T. Diversity of anthocyanin and proanthocyanin biosynthesis in land plants. Curr. Opin. Plant Biol. 2020, 55, 93–99. [Google Scholar] [CrossRef]
- Sinopoli, A.; Calogero, G.; Bartolotta, A. Computational aspects of anthocyanidins and anthocyanins: A review. Food Chem. 2019, 297, 124898. [Google Scholar] [CrossRef]
- Guo, X.L.; Shakeel, M.; Wang, D.L.; Qu, C.P.; Yang, S.M.; Ahmad, S.; Song, Z.J. Metabolome and transcriptome profiling unveil the mechanisms of light-induced anthocyanin synthesis in rabbiteye blueberry (Vaccinium ashei: Reade). BMC Plant Biol. 2022, 22, 223. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.A.; Dong, X.Y.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. Roy. Soc. B 2019, 374, 20190097. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lu, X.Y.; Xuan, Y.; Tang, F.; Wang, J.J.; Shi, D.; Fu, S.L.; Ren, J. Transcriptome analysis based on a combination of sequencing platforms provides insights into leaf pigmentation in Acer rubrum. BMC Plant Biol. 2019, 19, 240. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, D.O.N.; Yang, H.J.; Chen, M.B.; Zhang, W.T.; Zhang, L.Y. Metabolome and transcriptome sequencing analysis reveals anthocyanin metabolism in pink flowers of anthocyanin-rich tea (Camellia sinensis). Molecules 2019, 24, 1064. [Google Scholar] [CrossRef]
- Liu, X.W.; Wang, Y.H.; Shen, S.K. Transcriptomic and metabolomic analyses reveal the altitude adaptability and evolution of different-colored flowers in alpine Rhododendron species. Tree Physiol. 2022, 42, 1100–1113. [Google Scholar] [CrossRef]
- Wang, S.Z.; Li, Z.L.; Jin, W.B.; Fang, Y.P.; Yang, Q.F.; Xiang, J. Transcriptome analysis and identification of genes associated with flower development in Rhododendron pulchrum Sweet (Ericaceae). Gene 2018, 679, 108–118. [Google Scholar] [CrossRef]
- Li, Z.L.; Yang, Q.F.; Dong, X.; Zhu, Y.; Zhao, S.; Zhang, W.Y.; Wang, S.Z. Transcriptome analysis of flower color variation in five Rhododendron species (Ericaceae). Braz. J. Bot. 2021, 44, 685–695. [Google Scholar] [CrossRef]
- Nie, S.; Zhao, S.W.; Shi, T.L.; Zhao, W.; Zhang, R.G.; Tian, X.C.; Guo, J.F.; Yan, X.M.; Bao, Y.T.; Li, Z.C.; et al. Gapless genome assembly of azalea and multi-omics investigation into divergence between two species with distinct flower color. Hortic. Res.-Engl. 2023, 10, uhac241. [Google Scholar] [CrossRef]
- Ye, L.J.; Möller, M.; Luo, Y.H.; Zou, J.Y.; Zheng, W.; Wang, Y.H.; Liu, J.; Zhu, A.D.; Hu, J.Y.; Li, D.Z.; et al. Differential expressions of anthocyanin synthesis genes underlie flower color divergence in a sympatric Rhododendron sanguineum complex. BMC Plant Biol. 2021, 21, 204. [Google Scholar] [CrossRef]
- Polezhaeva, M.A.; Pimenova, E.A.; Tikhonova, N.A.; Korchagina, O.S. Plastid DNA diversity and genetic divergence within Rhododendron dauricum s.l. (R. dauricum s.s., R. ledebourii, R. sichotense and R. mucronulatum; Ericaceae). Plant Syst. Evol. 2018, 304, 763–774. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, H.Q.; Yuan, X.H.; Chen, Z.B.; Gao, J.B.; Teng, Y.; Yao, G.M. Grayanane diterpenoids from the leaves of Rhododendron dauricum. Biochem. Syst. Ecol. 2020, 89, 104009. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Y.; Li, F.Y.; Zhou, S.; Liu, G.L.; Yang, J.; Ye, W.B.; Wang, L. ‘Ao Xue’: A new Rhododendron cultivar. HortScience 2022, 57, 330–331. [Google Scholar] [CrossRef]
- Wang, Z.R.; Cui, Y.Y.; Vainstein, A.; Chen, S.W.; Ma, H.Q. Regulation of fig (Ficus carica L.) fruit color: Metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway. Front. Plant Sci. 2017, 8, 1990. [Google Scholar] [CrossRef] [PubMed]
- Kitteringham, N.R.; Jenkins, R.E.; Lane, C.S.; Elliott, V.L.; Park, B.K. Multiple reaction monitoring for quantitative biomarker analysis in proteomics and metabolomics. J. Chromatogr. B 2009, 877, 1229–1239. [Google Scholar] [CrossRef]
- Zhou, C.B.; Mei, X.; Rothenberg, D.O.N.; Yang, Z.B.; Zhang, W.T.; Wan, S.H.; Yang, H.J.; Zhang, L.Y. Metabolome and transcriptome analysis reveals putative genes involved in anthocyanin accumulation and coloration in white and pink tea (Camellia sinensis) flower. Molecules 2020, 25, 190. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef]
- Li, W.Z.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Liu, X.X.; Mei, W.B.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef]
- Li, J.W.; Ma, W.; Zeng, P.; Wang, J.Y.; Geng, B.; Yang, J.C.; Cui, Q.H. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.F.; Banf, M.; Dai, X.B.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, Y.; Cui, H.H.; Liu, J.W.; Wu, Y.Q.; Cheng, Y.; Xu, H.X.; Huang, X.X.; Li, S.T.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef]
- Du, H.; Lai, L.M.; Wang, F.; Sun, W.B.; Zhang, L.H.; Li, X.H.; Wang, L.S.; Jiang, L.H.; Zheng, Y.R. Characterization of flower coloration in 30 Rhododendron species via anthocyanin and flavonol identification and quantitative traits. Plant Biol. 2018, 20, 121–129. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, L.Y.; Wang, S.L.; Niu, X.Y. Analysis of anthocyanins and flavonols in petals of 10 Rhododendron species from the Sygera Mountains in Southeast Tibet. Plant Physiol. Bioch. 2016, 104, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Butelli, E.; Martin, C. Engineering anthocyanin biosynthesis in plants. Curr. Opin. Plant Biol. 2014, 19, 81–90. [Google Scholar] [CrossRef]
- Sun, X.B.; He, L.S.; Guo, Z.H.; Xiao, Z.; Su, J.L.; Liu, X.Q.; Zhou, H.M.; Li, C.; Gao, H.D. Comparative transcriptome analyses reveal genes related to pigmentation in the petals of a flower color variation cultivar of Rhododendron obtusum. Mol. Biol. Rep. 2022, 49, 2641–2653. [Google Scholar] [CrossRef]
- Wang, S.Z.; Huang, S.Y.; Yang, J.; Li, Z.L.; Zhang, M.J.; Fang, Y.P.; Yang, Q.F.; Jin, W.B. Metabolite profiling of violet, white and pink flowers revealing flavonoids composition patterns in Rhododendron pulchrum Sweet. J. Biosci. 2021, 46, 3. [Google Scholar] [CrossRef]
- Wang, M.; Chen, L.; Liang, Z.J.; He, X.M.; Liu, W.R.; Jiang, B.; Yan, J.Q.; Sun, P.Y.; Cao, Z.Q.; Peng, Q.W.; et al. Metabolome and transcriptome analyses reveal chlorophyll and anthocyanin metabolism pathway associated with cucumber fruit skin color. BMC Plant Biol. 2020, 20, 386. [Google Scholar] [CrossRef]
- Xia, X.; Gong, R.; Zhang, C.Y. Integrative analysis of transcriptome and metabolome reveals flavonoid biosynthesis regulation in Rhododendron pulchrum petals. BMC Plant Biol. 2022, 22, 401. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, M.F.; Wang, X.L.; Dai, J.; Zhang, X.; Zhang, Z.D.; Zhang, X.M.; Tang, M.; Tang, J.; Gong, J.Y.; et al. Transcriptome analysis of Rhododendron liliiflorum H. Lév. flower colour differences. Horticulturae 2023, 9, 82. [Google Scholar] [CrossRef]
- Chen, X.J.; Wang, P.J.; Gu, M.Y.; Hou, B.H.; Zhang, C.R.; Zheng, Y.C.; Sun, Y.; Jin, S.; Ye, N.X. Identification of PAL genes related to anthocyanin synthesis in tea plants and its correlation with anthocyanin content. Hortic. Plant J. 2022, 8, 381–394. [Google Scholar] [CrossRef]
- Lu, J.J.; Zhang, Q.; Lang, L.X.; Jiang, C.; Wang, X.F.; Sun, H.M. Integrated metabolome and transcriptome analysis of the anthocyanin biosynthetic pathway in relation to color mutation in miniature roses. BMC Plant Biol. 2021, 21, 257. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.F.; Zhao, D.H.; Zhang, J.Q.; Chen, J.S.; Li, J.L.; Weng, Z.; Rong, L.P. De novo transcriptome sequencing and anthocyanin metabolite analysis reveals leaf color of Acer pseudosieboldianum in autumn. BMC Genom. 2021, 22, 383. [Google Scholar] [CrossRef]
- Vikhorev, A.V.; Strygina, K.V.; Khlestkina, E.K. Duplicated flavonoid 3′-hydroxylase and flavonoid 3′, 5′-hydroxylase genes in barley genome. PeerJ 2019, 7, e6266. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Q.; Li, G.; Gu, L.Y.; Sun, Y.; Li, Z.Y.; Liu, J.R.; Wu, X.Q.; Dong, T.T.; Zhu, M.K. Comparative metabolomic and transcriptome analysis reveal distinct flavonoid biosynthesis regulation between petals of white and purple Phalaenopsis amabilis. J. Plant Growth Regul. 2020, 39, 823–840. [Google Scholar] [CrossRef]
- Shao, D.N.; Liang, Q.; Wang, X.F.; Zhu, Q.H.; Liu, F.; Li, Y.J.; Zhang, X.Y.; Yang, Y.L.; Sun, J.; Xue, F. Comparative metabolome and transcriptome analysis of anthocyanin biosynthesis in white and pink petals of cotton (Gossypium hirsutum L.). Int. J. Mol. Sci. 2022, 23, 10137. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhou, N.N.; Wang, Y.H.; Sun, S.Y.; Zhang, Y.; Ju, Z.G.; Yi, Y. Characterization and functional analysis of RdDFR1 regulation on flower color formation in Rhododendron delavayi. Plant Physiol. Biochem. 2021, 169, 203–210. [Google Scholar] [CrossRef]
- Jaakola, L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci. 2013, 18, 477–483. [Google Scholar] [CrossRef]
- Muhammad, N.; Luo, Z.; Yang, M.; Li, X.S.; Liu, Z.G.; Liu, M.J. The joint role of the late anthocyanin biosynthetic UFGT-encoding genes in the flowers and fruits coloration of horticultural plants. Sci. Hortic. 2022, 301, 111110. [Google Scholar] [CrossRef]
- Qian, R.J.; Ye, Y.J.; Hu, Q.D.; Ma, X.H.; Zhang, X.L.; Zheng, J. Metabolomic and transcriptomic analyses reveal new insights into the role of metabolites and genes in modulating flower colour of Clematis tientaiensis. Horticulturae 2023, 9, 14. [Google Scholar] [CrossRef]
- Ma, L.L.; Zhang, Y.P.; Cui, G.F.; Duan, Q.; Jia, W.J.; Xu, F.; Du, W.W.; Wang, X.N.; Li, X.; Chen, F.D.; et al. Transcriptome analysis of key genes involved in color variation between blue and white flowers of Iris bulleyana. BioMed Res. Int. 2023, 2023, 7407772. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tian, J.; Yao, Y.Y.; Zhang, J.; Song, T.T.; Li, K.T.; Yao, Y.C. Identification of leucoanthocyanidin reductase and anthocyanidin reductase genes involved in proanthocyanidin biosynthesis in Malus crabapple plants. Plant Physiol. Biochem. 2019, 139, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.P.; Vimolmangkang, S.; Soria-Guerra, R.E.; Korban, S.S. Introduction of apple ANR genes into tobacco inhibits expression of both CHI and DFR genes in flowers, leading to loss of anthocyanin. J. Exp. Bot. 2012, 63, 2437–2447. [Google Scholar] [CrossRef]
- Shi, M.F.; Xie, D.Y. Biosynthesis and metabolic engineering of anthocyanins in Arabidopsis thaliana. Recent. Pat. Biotechnol. 2014, 8, 47–60. [Google Scholar] [CrossRef]
- Qiu, L.K.; Zheng, T.C.; Liu, W.C.; Zhuo, X.K.; Li, P.; Wang, J.; Cheng, T.R.; Zhang, Q.X. Integration of transcriptome and metabolome reveals the formation mechanism of red stem in Prunus mume. Front. Plant Sci. 2022, 13, 884883. [Google Scholar] [CrossRef]
- Long, F.F.; Wu, H.R.; Li, H.E.; Zuo, W.W.; Ao, Q. Genome-wide analysis of MYB transcription factors and screening of MYBs involved in the red color formation in Rhododendron delavayi. Int. J. Mol. Sci. 2023, 24, 4641. [Google Scholar] [CrossRef]
- Fu, M.Y.; Yang, X.; Zheng, J.R.; Wang, L.; Yang, X.Y.; Tu, Y.; Ye, J.B.; Zhang, W.W.; Liao, Y.L.; Cheng, S.Y.; et al. Unraveling the regulatory mechanism of color diversity in Camellia japonica petals by integrative transcriptome and metabolome analysis. Front. Plant Sci. 2021, 12, 685136. [Google Scholar] [CrossRef]
- Zhou, S.F.; Chen, J.; Lai, Y.S.; Yin, G.H.; Chen, P.L.; Pennerman, K.K.; Yan, H.D.; Wu, B.C.; Zhang, H.; Yi, X.F.; et al. Integrative analysis of metabolome and transcriptome reveals anthocyanins biosynthesis regulation in grass species Pennisetum purpureum. Ind. Crops Prod. 2019, 138, 111470. [Google Scholar] [CrossRef]
- Deng, J.; Li, J.J.; Su, M.Y.; Lin, Z.Y.; Chen, L.; Yang, P.F. A bHLH gene NnTT8 of Nelumbo nucifera regulates anthocyanin biosynthesis. Plant Physiol. Biochem. 2021, 158, 518–523. [Google Scholar] [CrossRef]
- Zhong, Y.D.; Chen, C.H.; Gong, X.; Luan, X.Y.; Wu, Z.X.; Li, H.H.; Liu, Q.L.; Xu, M.; Yu, F.X. Transcriptome and metabolome analyses reveal a key role of the anthocyanin biosynthetic pathway cascade in the pigmentation of a Cinnamomum camphora red bark mutant (‘Gantong 1’). Ind. Crops Prod. 2022, 175, 114236. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene-ID | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| 18S rRNA | GTAGGCGGTGAATCGGGTTG | TGGCGTTCCTTCGTAGATGGTT |
| α-actin | TGACTATGAGCAAGAGCTGGAGA | CAAAGATGGCTGGAAGAGGAC |
| Transcript/8857 | CCATACCCTGCCTCGTTC | AATTCGGGCTTTCCTTGC |
| Transcript/26078 | GCCGATTCTCAGCATCAC | CAATTCCTCCACAAACCC |
| Transcript/13928 | CCCGATTACTACTTTCGTATC | CGTCTATTGCGTGTTTGAG |
| Transcript/20082 | TCCGTTGAGTTCTTTAGG | GTTCTTGATTACCGCTTT |
| Transcript/18962 | TGGATGCGGGTCTGATTT | CCAACCTGGTCCTGAAGC |
| Transcript/18781 | ATAGAGGGCGGGATGAAA | GCTTGTAGGTAAGGCAGGTT |
| Transcript/19488 | GTCAAGAGGCTGGTGTTC | TCGCCAAATCGTAGATAG |
| Transcript/19757 | TTTTAGGTCGTGGGTTAG | CAGACTCCCAAATGCTAC |
| Transcript/15418 | ACCGTCACTTTCTCCTTCT | GACCATAAACGAGACCCAC |
| Transcript/14301 | TGCGTTGTCGTGGTTTGA | ATGCGAGCCCATTTGTTA |
| Transcript/19510 | AGACAACAATAGCGACATA | ATCTTAGCCAATTCTACATC |
| Transcript/4872 | TGTGCCAATGGTAGTATGT | TTTGTTAATGGGTTAGGG |
| Transcript/10990 | CAAGGGTATGGTGGAATC | GGAGTAGAGGGCTTATCAA |
| Transcript/12417 | GAAAGGGAGTGAGGTTGG | ATGCCTGAGAAGGAGTAGAG |
| Transcript/15129 | GAGACGATGGTCCAGTTCA | CCCAACCTGCTCATACTCA |
| Sample | Total Reads | Mapped Reads Percentage (%) | Uniquely Mapped Reads Percentage (%) | Multiple Mapped Reads Percentage (%) | |
|---|---|---|---|---|---|
| R. dauricum var. albiflorum | Replicate1 | 25,896,223 | 68.65 | 47.27 | 21.38 |
| Replicate2 | 30,609,347 | 68.54 | 48.00 | 20.54 | |
| Replicate3 | 26,848,646 | 68.82 | 48.37 | 20.45 | |
| R. dauricum | Replicate1 | 24,159,942 | 68.10 | 47.78 | 20.32 |
| Replicate2 | 27,634,348 | 68.18 | 47.91 | 20.27 | |
| Replicate3 | 26,275,262 | 68.03 | 47.68 | 20.35 | |
| Transcription Factor | Gene-ID | log2(Fold Change) | Up/Down |
|---|---|---|---|
| MYB | Transcript/1227 | −1.52 | Down |
| Transcript/14301 | −1.05 | Down | |
| Transcript/18795 | 4.21 | Up | |
| Transcript/19510 | −1.08 | Down | |
| Transcript/22613 | 3.74 | Up | |
| Transcript/24760 | 4.24 | Up | |
| Transcript/2607 | −1.42 | Down | |
| Transcript/3024 | 4.34 | Up | |
| Transcript/3462 | −1.52 | Down | |
| Transcript/4872 | 2.02 | Up | |
| Transcript/6971 | −11.70 | Down | |
| Transcript/7286 | −1.38 | Down | |
| Transcript/7388 | 2.30 | Up | |
| Transcript/8473 | 2.92 | Up | |
| Transcript/965 | 1.28 | Up | |
| bHLH | Transcript/10990 | −1.24 | Down |
| Transcript/12417 | 1.09 | Up | |
| Transcript/12837 | 9.35 | Up | |
| Transcript/15129 | 1.57 | Up | |
| Transcript/16447 | −3.04 | Down | |
| Transcript/16636 | −12.21 | Down | |
| Transcript/19483 | −1.47 | Down | |
| Transcript/7084 | 10.83 | Up | |
| Transcript/7831 | 1.33 | Up | |
| Transcript/9298 | −5.96 | Down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, H.; Jin, X.; Zhang, Y.; Lin, L. Anthocyanin Metabolite and Transcriptome Sequencing Analysis Reveals White Flowers in Rhododendron dauricum var. albiflorum. Horticulturae 2023, 9, 1071. https://doi.org/10.3390/horticulturae9101071
Mu H, Jin X, Zhang Y, Lin L. Anthocyanin Metabolite and Transcriptome Sequencing Analysis Reveals White Flowers in Rhododendron dauricum var. albiflorum. Horticulturae. 2023; 9(10):1071. https://doi.org/10.3390/horticulturae9101071
Chicago/Turabian StyleMu, Huaizhi, Xuhong Jin, Yue Zhang, and Lin Lin. 2023. "Anthocyanin Metabolite and Transcriptome Sequencing Analysis Reveals White Flowers in Rhododendron dauricum var. albiflorum" Horticulturae 9, no. 10: 1071. https://doi.org/10.3390/horticulturae9101071