Abstract
The rapid ripening of soursop (Annona muricata L.) fruits is owing to its high respiration rate. Several enzymes affect the fruit cell wall in this process, resulting in the depolymerization of pectin primarily in the homogalacturonan. The main group of enzymes affecting the pectin content of soursop fruits include polygalacturonase (PG), pectate lyase (PL), pectin methylesterase (PME), and PME inhibitors (PMEis). In this study, pectin-degrading enzymes were analyzed using bioinformatic tools to uncover the gaps in our knowledge of this fruit. In this context, 67 genes encoding PG, 33 PL, 58 PME, and 39 PMEi isoenzymes were found. These genes were categorized into several families based on the results of phylogenetic analysis. Regarding the analysis of gene expression, a total of 25 were identified as differentially expressed genes (DEGs) in PG, while 3, 21, and 15 were found for PL, PME, and PMEis, respectively. Likewise, functional enrichment analysis demonstrated that the DEGs are involved in the modification of the cell wall, specifically in the degradation of pectin. On the other hand, gene co-expression networks revealed that the genes PG32 and PG35 affect the expression of PGs, as well as PL19 of the PL family, PME20, PME32, and PME35 of the PME family, and PMEi04 of the PMEi family. This suggests that they have a significant impact on the softening of soursop fruits.
1. Introduction
Mexico is the primary global producer of soursop fruit (Annona muricata L.), with a reported production of 34,437.03 t in the year 2022. Notably, around 78% of this total production was cultivated in Nayarit [1]. However, the soursop is a climacteric fruit due to its high respiration rate and ethylene production, which causes rapid maturation, thereby limiting its commercialization [2]. The ripening of fleshy fruits is a complex process that involves a series of physiological and biochemical changes, such as the modification of color due to the accumulation of pigments and the degradation of chlorophyll, the formation of flavor as organic acids and sugars accumulate along with volatile compounds, as well as softening involving cell wall degradation, whereby pectin is solubilized and depolymerized [3,4]. Pectins are structurally very complex polysaccharides that play an important role in cell adhesion. These are mainly composed of homogalacturonans (HGs), xylogalacturonan (XGA), rhamnogalacturonan-I (RG-I), and rhamnogalacturonan-II (RG-II) [5]. This pectin matrix undergoes modifications that impact the mechanical properties of the cell wall, primarily in HG, in which a group of enzymes is involved [6,7]. One of the enzymes present in this group is pectin methylesterase (PME), which facilitates the de-esterification process of galacturonic acid residues located in the primary structure of HG. This enzymatic reaction results in the production of carboxyl groups and the liberation of methanol within the cell wall. Following de-esterification, two things may happen; first, the de-esterified HG can form bridges with calcium ions to form a structure called an “eggbox”, causing rigidity in the cell wall, or HG can become a target for enzymes, such as polygalacturonase (PG) and pectate lyase (PL) [8]. PG hydrolyzes the α-1,4 bonds between the galacturonic acid molecules adjacent to the esterified HG backbone [9]. In contrast, PL cleaves the α-1,4 bonds of galacturonic acid via a β-elimination process [10]. However, it is important to note that the activity of PME (which plays a role in the level of methyl esterification of HG, impacting both cell wall hardening and disintegration) is primarily regulated via endogenous proteins known as PME inhibitors (PMEis) [11]. In this context, biochemical and molecular studies have been carried out that confirmed the role of these enzymes in the fruit softening of the Annonaceae family, such as saramuyo (Annona squamosa L.) and atemoya (Annona cherimola Mill. × Annona squamosa L.), reporting a high enzymatic activity and gene expression of PG, PL, and PME in mature fruits [12,13,14]. In soursop, efforts have been made to understand fruit softening, and only a few reports have focused on the expression analysis of the PL, PG, and PME genes, finding higher expression levels on day 6 compared to day zero of storage [15,16]. The identification and characterization of the members belonging to the PG, PL, PME, and PMEi gene families in soursop fruits have not been performed thus far. Consequently, the expression patterns of these genes remain unknown. Therefore, it is imperative to analyze the genes associated with pectin degradation in soursop fruits. This endeavor is crucial for extending the fruit’s shelf life and facilitating its entry into international markets. Therefore, the objective of this work was to identify the members of the families of the PL, PG, PME, and PMEi genes, their phylogenetic relationship, as well as evaluate their gene expression and functional categorization during the ripening of the soursop fruit through bioinformatic analysis.
2. Materials and Methods
2.1. Gene Identification and Phylogenetic Analysis
In a previous study, our research group conducted de novo assembly of soursop fruits at different stages of ripening. This analysis resulted in the identification of a total of 95,832 unigenes. Among these unigenes, only 22,319 were found to possess protein domain annotation based on the Pfam database [15]. This transcriptome (BioProject ID number PRJNA804904), along with the freely accessible database named the Annomics Database http://perseo.uan.mx/bioinformatica/annomicsdatabase (accessed on 10 July 2023) [15], was utilized to identify genes encoding for the enzymes PG, PL, PME, and PMEi, considering the Pfam as the main criterion: PF00295 for PG, PF12708, PF00544, and PF04431 for PL, PF01095 for PME, and PF04043 for PMEi. Subsequently, a nucleotide sequence alignment of these genes was conducted using the MEGA v11.0 program with the MUSCLE statistical method, selecting the default parameters [17]. Once the sequences were aligned, a phylogenetic tree was built for each enzyme family (PG, PL, PME, and PMEi) using the IQ_TREE v2.2.0 program at http://www.iqtree.org/ (accessed on 17 August 2023), with the distance model based on the neighbor-joining algorithm and considering a Bootstrap of 1000 replicates [18]. Finally, the figures were performed and edited in iTOL v6.0 at https://itol.embl.de/ (accessed on 19 August 2023) [19].
2.2. Differential Expression Analysis
Differential gene expression analysis was performed from a RSEM (RNA-seq by expectation maximization) data matrix using the DESeq2 package in Rstudio [20]. In order to identify the up- and down-regulated genes, three pairwise comparisons against day zero (physiological maturity) were performed (day 3 vs. 0, day 6 vs. day 0, and day 9 vs. day 0). The genes that presented a Log2 fold change > 1 and a Padj < 0.05 were considered differentially expressed genes (DEGs). Finally, the annotated genes for PG, PL, PME, and PMEi were plotted on a heat map through the TBtools program [21].
2.3. Mapping to the Cherimoya (Annona cherimola) Genome
The sequences of the DEGs were mapped to the cherimoya genome due to it belonging to the Annonaceae family, and due to its genome being freely available and having high-quality sequences at https://ihsmsubtropicals.uma.es/ (accessed on 1 September 2023) [22]. Indeed, in order to identify the possible chromosomal location of each gene, BLAST analysis was performed, with the cut-off parameters percentage of identity > 90% and e-value < 0.05.
2.4. Functional Enrichment Analysis
A functional enrichment of the DEGs was performed using the Blast2GO program [23] to obtain the gene ontology (GO) terms associated with the biological process, molecular function, and cellular component in the plant cell wall. Likewise, the annotation in the Kyoto Encyclopedia of Genes and Genomes (KEGG) of the DEGs was conducted. Following this step, the metabolic pathway was determined according to the report in the KEGG database at https://www.genome.jp/kegg/ (accessed on 1 September 2023) using the Arabidopsis thaliana organism.
2.5. Gene Co-Expression Networks
To identify the genes with a significant association between expression levels, a correlation matrix was calculated between the enriched DEGs using Pearson’s correlation coefficient and a significance level of p < 0.05. The co-expression networks were then constructed using the undirected model with the igraph package in Rstudio, removing the edges that presented an r < 0.8. Likewise, the graph was converted into a minimum spanning tree (MST) based on Prim’s algorithm to only obtain a subset of the edges that connects all the vertices without any cycles. The nodes represented the DEGs, and the edges indicated the positive or negative correlation between the DEGs.
3. Results
3.1. Phylogenetic and Gene Differential Expression Analysis
Fifty-eight isoenzymes containing the PME functional domain PF01095 were identified. In terms of the phylogenetic analysis, the PME isozymes were classified into 14 families (F), with F5, F11, and F14 containing the greatest number of isoenzymes (Figure 1a). It was observed that there were independent genes that presented branch length values greater than 1.0, indicating that they are evolutionarily distinct from the remaining genes (F1, F2, F4, F6, and F9). In relation to the differential expression analysis, a total of 21 DEGs were identified, exhibiting expression patterns across all days of storage analyzed (Figure 1b). Among the DEGs that were identified, a total of 10 genes were observed to be up-regulated. Notably, PME32 and PME35 exhibited the highest levels of expression, with a 26- and 23-fold increase than those observed on day zero, respectively. In contrast, 11 down-regulated genes were found; PME28 and PME08 exhibited the lowest levels of expression, with a 7- and 5-fold decrease in their gene expression compared that under day zero, respectively.
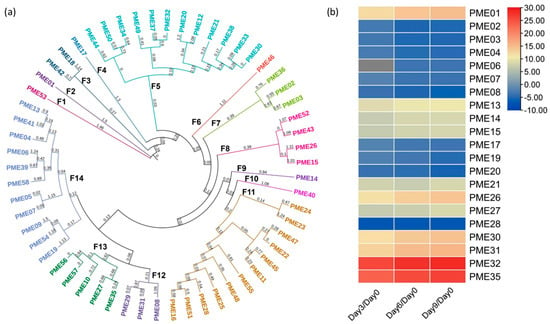
Figure 1.
(a) Phylogenetic analysis using the neighbor-joining method of PME genes, where “F” represents the number of families in which they were grouped. Likewise, the color of the genes indicates how they were grouped within their respective family according to the analysis. The consecutive number was assigned to each gene to identify them. (b) Differential expression analysis of the PME genes. The color scale indicates the Log2 fold change in the DEGs (Padj < 0.05); the up-regulated genes represent the levels from zero to thirty, while the down-regulated genes represent the levels from zero to minus ten, according to the color scale ranging from red to blue.
Concerning the PMEis, 39 isoenzymes with the functional domain PF04043 were identified as PME inhibitors. According to the phylogenetic analysis, the soursop PMEi isoenzymes were classified into five families, with the majority of these isoenzymes linked to the F5 family. Furthermore, F1, F2, and F3 were independent families (Figure 2a). Subsequently, fifteen DEGs were found, and only four were up-regulated, namely PMEi06, PMEi17, PMEi29, and PMEi32. Indeed, three of them were detected in F5, as shown in Figure 2b. The expression levels of these genes showed a 6-fold increase compared to day zero. The remaining down-regulated genes were located in different families, with F2 constituting a highly repressed independent family, as the genes of this family were repressed, showing a 10-fold decrease in comparison with day zero.
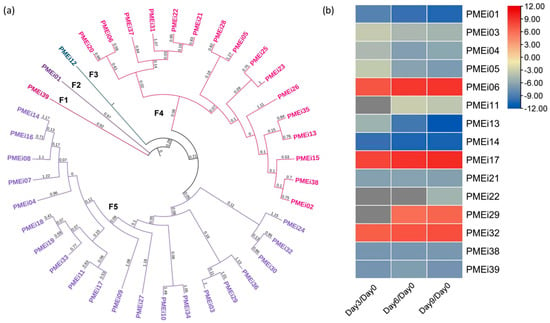
Figure 2.
(a) Phylogenetic analysis using the neighbor-joining method of PMEi genes, where “F” represents the number of families in which they were grouped. Likewise, the color of the genes indicates how they were grouped within their respective family according to the analysis. The consecutive number was assigned to each gene to identify them. (b) Differential expression analysis of the PMEi genes. The color scale indicates the Log2 fold change in the DEGs (Padj < 0.05); the up-regulated genes represent the levels from zero to twelve, while the down-regulated genes represent the levels from zero to minus twelve, according to the color scale ranging from red to blue.
On the other hand, 67 PG isoenzymes with the functional domain PF00295 corresponding to family 28 of glycosyl hydrolases were identified. Based on the results of the phylogenetic analysis, PGs were classified into eight distinct families, as depicted in Figure 3a. The F8 family exhibited the largest number of members, while F1 was the only independent family. These isoenzymes contained 25 DEGs, of which 20 were up-regulated (Figure 3b). PG15, PG32, and PG35, which are all located in F4, were more up-regulated during all the days analyzed, with an 11-fold increase in gene expression compared to day zero. In contrast, PG02, PG48, and PG20 were found to be down-regulated around 3-fold compared to day zero in F6, F3, and F8, respectively.
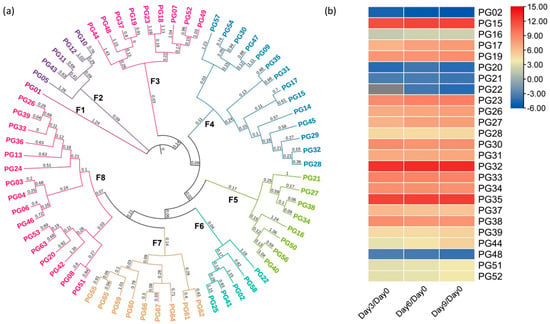
Figure 3.
(a) Phylogenetic analysis using the neighbor-joining method of PG genes, where “F” represents the number of families in which they were grouped. Likewise, the color of the genes indicates how they were grouped within their respective family according to the analysis. The consecutive number was assigned to each gene to identify them. (b) Differential expression analysis of the PG genes. The color scale indicates the Log2 fold change in the DEGs (Padj < 0.05); the up-regulated genes represent the levels from zero to fifteen, while the down-regulated genes represent the levels from zero to minus six, according to the color scale ranging from red to blue.
Finally, 21 PL isoenzymes with the functional domain PF12708 were identified, corresponding to proteins of the pectate lyase superfamily. Additionally, 11 PL isoenzymes with the PF00544 domain were found, and one isoenzyme with the PF04431 domain was found to correspond to the PL N-terminal. These 33 isoenzymes were grouped into seven families according to the phylogenetic analysis (Figure 4a). Nonetheless, it is worth noting that only three DEGs from the F7 family were detected, as shown in Figure 4b. Among these DEGs, two genes (PL12 and PL19) exhibited a 3-fold increase in gene expression for all days compared to day zero.
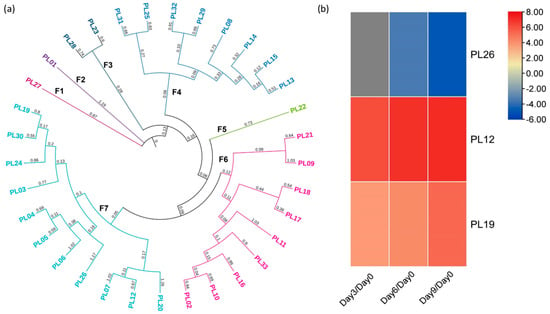
Figure 4.
(a) Phylogenetic analysis using the neighbor-joining method of PL genes, where “F” represents the number of families in which they were grouped. Likewise, the color of the genes indicates how they were grouped within their respective family according to the analysis. The consecutive number was assigned to each gene to identify them. (b) Differential expression analysis of the PL genes. The color scale indicates the Log2 fold change in the DEGs (Padj < 0.05); the up-regulated genes represent the levels from zero to eight, while the down-regulated genes represent the levels from zero to minus, according to the color scale ranging from red to blue.
3.2. Mapping into the Cherimoya Genome
The BLAST analysis of the PG, PL, PME, and PMEi genes revealed that 50 of the 64 DEGs analyzed were homologous to the cherimoya genes, as they presented an identity of 90–96% with an e-value close to zero (Supplementary Materials, Table S1). In the case of PG, 20 DEGs that matched the cherimoya genes were identified; PG15, PG32, and PG35, which were predominantly up-regulated, were located on chromosome 7. Regarding PL, chromosomes six, seven, and three contained the PL26, PL12, and PL19 genes, respectively. PME contained 13 DEGs, including PME32, which is located on chromosome one and is one of the most up-regulated genes. Finally, 14 DEGs were identified for the PMEis, including PMEi06, PMEi17, and PMEi32 (significantly up-regulated). They were all located on chromosome one (Supplementary Materials, Table S1).
3.3. Functional Enrichment Analysis
This analysis revealed more than 10 significant GO terms related to cell wall modification. The GO annotation of these terms was related to the organization, modification, and biogenesis of the cell wall, the enzymatic activity of PG and PME, as well as the regulatory and inhibitory activity of enzymes, as shown in Table 1.

Table 1.
Enriched GO terms associated with the cell wall.
Similarly, it was observed that several significant DEGs were associated with multiple GO terms. For instance, PME35, PME32, PME28, and PME08 exhibited six GO terms. Additionally, PMEi32, PMEi29, and PMEi17 were linked to four terms, while PG48, PG35, PG32, PG20, and PG15 showed two GO terms (Table 2).
Table 2.
GO terms of enriched genes associated with a biological process, molecular function, and cellular component.
On the other hand, the KO terms of the DEGs were compared with the KEGG database, resulting in three predominant KO terms (KO: KO1051, KO: KO1728, and KO: KO3083) corresponding to the PME, PL, and PG genes, respectively. These KO terms were positioned in the carbohydrate metabolism pathway (ath01100), through the pentose and glucuronate interconversion pathway (ath00040), specifically in the pectin degradation module (ath_M00081), as shown in Figure 5.
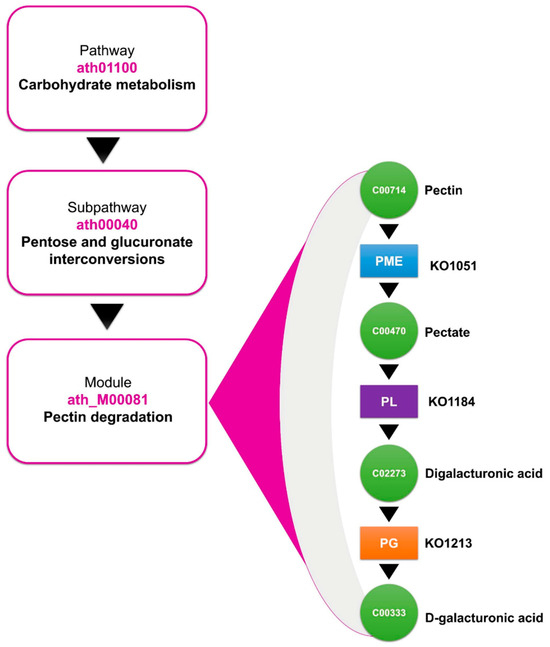
Figure 5.
KEGG annotations of the DEGs of the PG, PL, and PME enzymes, located in the pectin degradation module through the pentose and glucuronate interconversion pathway, situated in carbohydrate metabolism, according to the Kyoto Encyclopedia of Genes and Genomes.
3.4. Gene Co-Expression Network
An MST network containing all vertices with the minimum length was inferred from the Log2 fold change of the enriched DEGs for each enzyme analyzed (Figure 6). Highly associated genes are connected and reveal hierarchical relationships as evidence of an undirected and random structure. According to Figure 6, the PME genes were distributed in three groups, which correspond to a subtree of the MST. The PME network comprises a total of 20 nodes and 19 connections, with PME27 and PME13 showing two strong interactions. A negative correlation was observed between PME13 and PME19, which suggests that when PME13 was up-regulated, PME19 was down-regulated. In turn, PME19 presented five positive and two negative correlations, of which six coincided with the differential expression analysis since the genes with a positive correlation were found to be down-regulated. On the other hand, in the case of PME27, PME31 presented a positive correlation and also presented a negative correlation with PME20, which interacts positively and negatively with eight genes and coincides with their expression. Regarding the PMEis, Figure 7 shows that they were divided into two groups. This network consists of 14 nodes and 12 connections; it was observed that PMEi39 and PMEi21 had no correlation with the remaining nodes, but a positive correlation with one another. However, PMEi04 maintained 11 positive and negative interactions, most of which were positive, suggesting that this gene probably influences the expression of the remaining differentially expressed PMEi genes.
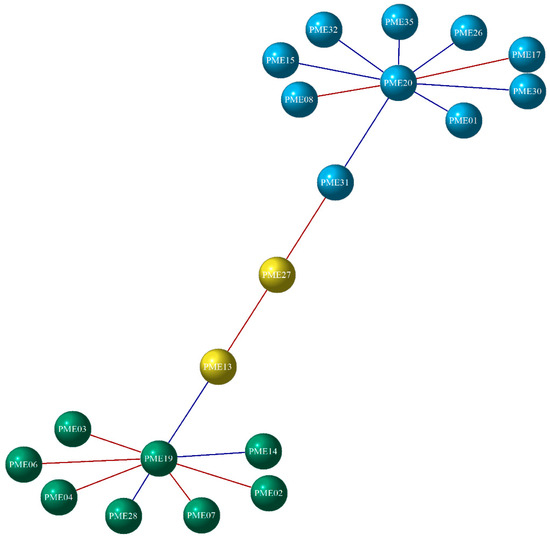
Figure 6.
The gene co-expression network of PME. DEGs are represented by the nodes (circles), with each group distinguished by a unique color (blue, yellow, and green). Red lines indicate a positive correlation, while blue lines indicate a negative correlation.
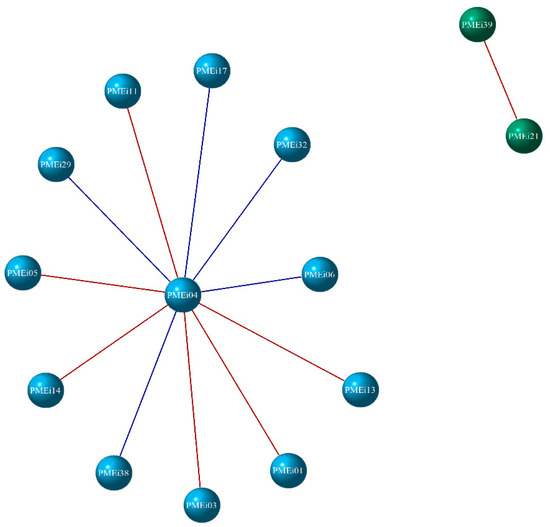
Figure 7.
The gene co-expression network of PMEis. DEGs are represented by nodes (circle), with each group distinguished by a unique color (sky blue and green). Red lines indicate a positive correlation, while blue lines indicate a negative correlation.
On the other hand, according to Figure 8, PG was dispersed into four groups. The PG network contains 25 nodes and 24 connections. It can be seen that each group was made up of six genes that are mostly positively correlated with each other. The yellow colored group contained up-regulated genes with a positive correlation with each other; the sky-blue group presented PG52 as the main gene that interacts with the remaining genes positively and negatively. Similarly, in the green group, it was observed that PG51 was correlated with the rest, mainly negatively. In the blue group, one gene depended on another, suggesting a cascading-type correlation.
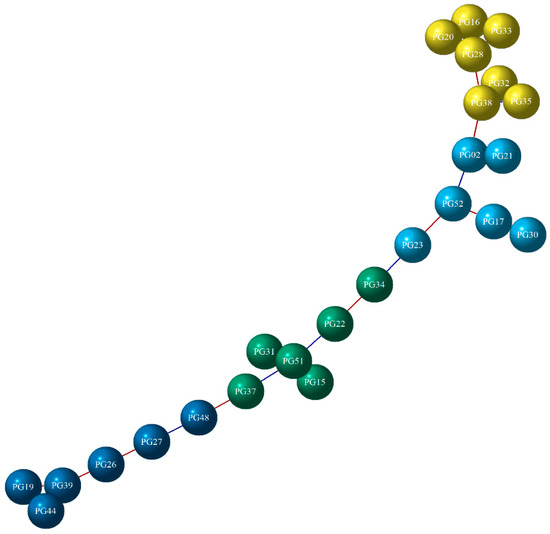
Figure 8.
The gene co-expression network of PG. DEGs are represented by nodes (circles), with each group distinguished by a unique color (yellow, sky blue, green, and blue). Red lines indicate a positive correlation, while blue lines indicate a negative correlation.
The DEGs of PL did not form clusters but instead correlated with one another. Its network consists of three nodes and two connections, with PL19 being positively and negatively correlated with PL26 and PL12, respectively (Figure 9).
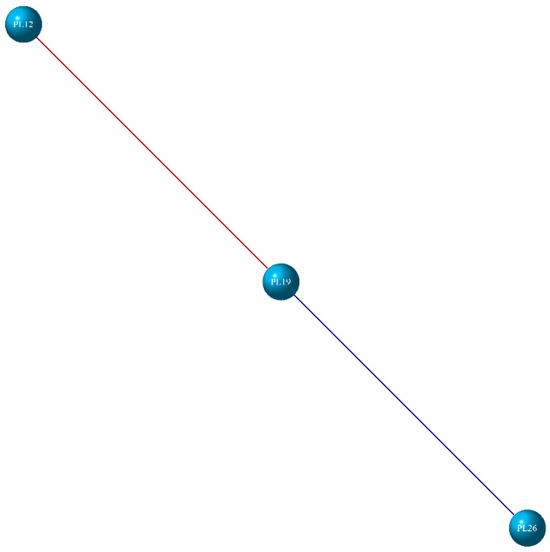
Figure 9.
The gene co-expression network of PL. DEGs are represented by nodes (circles). Red lines indicate a positive correlation, while blue lines a indicate negative correlation.
4. Discussion
4.1. PME and PMEis
Fifty-eight PME isoenzymes were found in soursop fruits during ripening; this is a similar number to the ones reported in Arabidopsis thaliana with 66 genes, in oranges (Citrus x sinensis) with 53 genes, in wild strawberries (Fragaria vesca) with 54 genes, and in Annona montana with 45 genes [24,25,26,27]. PME participates in several physiological processes involved in the vegetative and reproductive development of plants, as well as in responses to various types of biotic and abiotic stresses [28]. During fruit development, PME removes a methyl group from highly esterified pectins, causing them to form bridges with Ca2+ ions, thereby enhancing cell adherence and, consequently, the rigidity and mechanical properties of the fruit’s cell wall [7]. However, once fruit ripening begins, PME exerts another function. When pectin is minimally esterified, PME provides a cleavage site in homogalacturonans for pectin-degrading enzymes such as PG [11]. In this regard, it is probable that the PME genes that were differentially expressed, such as PME32 and PME35, which were strongly up-regulated, are involved in cell wall alterations during ripening and, hence, fruit softness. GO terms define the molecular functions of genes, as well as their role in cellular processes and their location in cellular components [25]. In this sense, the PME genes showed a GO term associated with cell wall modification, supporting its function. However, regarding the gene expression networks, PME20 is important, as it affects the expression of PME32 and PME35 as they are negatively correlated, suggesting that PME20 must remain down-regulated for PME32 and PME35 to be up-regulated and execute their functions.
On the other hand, due to the dual function of PME, it is important to control their activity in the fruit ripening process; therefore, there are PME inhibitors (PMEis), which bind to PME and regulate its activity [5]. Regarding the PMEis, 39 isoenzymes were found, and these genes have been reported in other plants, such as Arabidopsis thaliana with 78 genes and in white pear (Pyrus bretschneideri) with 42 genes [29,30]. The function underlying the PMEis mainly occurs during the plant growth process; their overexpression has been observed, for example, in seed germination, as well as in flowering in Arabidopsis thaliana [29]. Its participation has also been reported during the development of fruits, such as grapes (Vitis vinifera L.), oranges, and tomatoes (Solanum lycopersicum L.) [8,26,31]. In this context, the possible reason for why only four PMEi genes were positively expressed is that the function of PME during maturation is to modify the cell wall, and since it is a genetically programmed process [32], the up-regulation of PMEis is not extremely necessary, and their greatest activities can only be found in the initial stages of fruit growth. Likewise, the PMEi genes showed a GO term associated with the regulatory and inhibitory activity of enzymes.
In terms of the co-expression networks, the up-regulated genes (PMEi06, PMEi17, PMEi29, and PMEi32) affected the expression of PMEi04, suggesting that it is important, as if it is repressed, the rest will be directly affected. Hence, analyzing these five genes would be useful for future research.
4.2. PG
In our analysis, 67 PG isoenzymes were found, and 55 PG genes have been reported in maize (Zea mays L.), 45 genes in sweet cherry (Prunus avium), 24 in mango (Mangifera indica L.), and 30 genes in A. montana [27,33,34,35]. PG presented the highest number of DEGs compared to the rest of the enzymes analyzed, most of which were up-regulated. In turn, the expression levels had increased according to the days of ripening, as PG is one of the enzymes that is mostly up-regulated in maturation and is responsible for softening since it hydrolyzes the homogalacturonan chains of de-esterified pectins that were previously de-esterified by PME [36]. This coincides with that reported in atemoya fruits, indicating an increase in the expression levels of most of the genes that code for PG when ripened for consumption [14]. Even though the majority of the DEGs were up-regulated, PG32 and PG35 demonstrated the greatest increase in gene expression. These genes showed GO terms such as polygalacturonase activity, so we can infer that these genes degrade the cell wall. In the co-expression networks, most of the genes were associated in a cascade manner, where some depend positively or negatively on others. Nevertheless, PG32 and PG35 were associated in the same group and with a positive correlation, where the rest of the genes are also up-regulated, which probably means that this group of enzymes impacts the hydrolytic process of the HG of pectins.
4.3. PL
Concerning the PL genes, twenty-one isoenzymes were found, of which only three were expressed, two up-regulated and one down-regulated. This coincides with that reported in mangoes, since they identified eighteen PL genes in the transcriptional analysis during mango ripening, of which only two were differentially expressed, one repressed, and the other induced [33]. Lastly, it is possible that some enzymes are up- or down-regulated at different ripening stages, as soursop is a multiple fruit in which the ovaries, being apocarpic, do not fertilize uniformly, meaning that different times could have elapsed in the formation of the fruit from different ovaries [37]. On the other hand, based on gene mapping analysis, it has been shown that each gene is located on distinct chromosomes, suggesting that their genomic positioning may correspond to diverse functions throughout various stages of fruit development.
5. Conclusions
In this study, genes associated with cell wall modification, particularly pectin degradation, were identified. Fifty-eight genes coding for PME isoenzymes were found, of which only twenty-one were differentially expressed. Among these genes, particular attention might be directed towards the PM32 and PME35 genes due to their predominantly up-regulated expression patterns and enrichment GO terms associated with cell wall modifications. Likewise, it is important to mention PME20, which regulates the expression of PME32 and PME35 according to the co-expression gene networks. Furthermore, a total of 39 genes that encode PMEi isoenzymes were identified, of which 15 DEGs were shown. Among them, four up-regulated genes (PMEi06, PMEi17, PMEi29, and PMEi32) were found, which might be involved in the regulatory and inhibitory activity of enzymes. Likewise, 67 genes that encode PG isoenzymes were identified. Out of the set of PG genes examined, a total of 25 genes exhibited differential expression, with the bulk of these genes being up-regulated. Notably, the genes PG32 and PG35 had the highest degree of up-regulation. The aforementioned genes exhibit enrichment and, as indicated by the co-expression network, they may engage in beneficial interactions with another cluster of genes. Finally, thirty-nine genes encoding PL isoenzymes were identified, of which only three genes were differentially expressed. Based on these results, we identified key genes that degrade pectin during soursop fruit ripening. Future studies will be carried out to analyze the expression of genes that code for the PG, PL, PME, and PMEis in soursop fruits.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/horticulturae9101150/s1, Table S1: BLAST comparison between soursop and cherimoya genes.
Author Contributions
Conceptualization, formal analysis, writing—original draft, L.A.D.-R.; writing—review and editing, investigation, V.A.O.-J. and G.B.-V.; data curation, P.U.B.-R.; investigation, methodology, R.B.-M. and J.E.B.-L.; resources, project administration, funding acquisition, G.B.-V. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to thank Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCYT) for the financial support by the grant Ciencia Básica y/o Ciencia de Frontera Modalidad Paradigmas y Controversias de la Ciencia, grant number 319996: “Análisis integral de datos transcriptómicos y metabolómicos asociados a la calidad de los frutos de guanábana (Annona muricata L.) durante almacenamiento poscosecha”.
Data Availability Statement
The data presented in this study are available within the article.
Acknowledgments
The first author thanks the scholarship granted by CONAHCYT for their Ph.D. studies, grant number 349748.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Servicio de Información Agroalimentaria y Pesquera (SIAP). Available online: https//nube.siap.gob.mx/cierreagricola/ (accessed on 15 August 2023).
- Nayak, A.; Hegde, K. A comprehensive review on the miracle nature of Annona muricata. J. Pharm. Sci. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Dos Santos, R.S.; Arge, L.W.; Costa, S.I.; Machado, N.D.; de Mello-Farias, P.C.; Rombaldi, C.V.; de Oliveira, A.C. Genetic regulation and the impact of omics in fruit ripening. Plant Omics 2015, 8, 78–88. [Google Scholar]
- Omboki, R.B.; Wu, W.; Xie, X.; Mamadou, G. Ripening genetics of the tomato fruit. Int. J. Agric. Crop Sci. 2015, 8, 567–572. [Google Scholar]
- Zhang, Q.; Pu, T.; Wang, Y.; Bai, Y.; Suo, Y.; Fu, J. Genome-wide identification and expression analysis of the PME and PMEi gene families in Diospyros kaki: A bioinformatics study. Horticulture 2022, 8, 1159. [Google Scholar] [CrossRef]
- Philippe, F.; Pelloux, J.; Rayon, C. Plant pectin acetylesterase structure and function: New insights from bioinformatic analysis. BMC Genom. 2017, 18, 456. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Chan, A.; Jung, M.; Lee, Y. Recent advances in understanding the roles of pectin as an active participant in plant signaling networks. Plants 2021, 10, 1712. [Google Scholar] [CrossRef]
- Wen, B.; Zhang, F.; Wu, X.; Li, H. Characterization of the tomato (Solanum lycopersicum) pectin methylesterase: Evolution, activity of isoforms and expression during fruit ripening. Front. Plant Sci. 2020, 11, 238. [Google Scholar] [CrossRef]
- Huang, W.; Chen, M.; Zhao, T.; Han, F.; Zhang, Q.; Liu, X.; Jiang, C.; Zhong, C. Genome-wide identification and expression analysis of polygalacturonase gene family in kiwifruit (Actinidia chinensis) during fruit softening. Plants 2020, 9, 327. [Google Scholar] [CrossRef] [PubMed]
- Al Hinai, T.Z.S.; Vreeburg, C.L.M.; Murray, L.; Sadler, I.H.; Fry, S.C. Fruit softening: Evidence for pectate lyase action in vivo in date (Phoenix dactylifera) and rosaceous fruit cell walls. Ann. Bot. 2021, 128, 511–525. [Google Scholar] [CrossRef]
- Wormit, A.; Björn, U. The multifaceted role of pectin methylesterase inhibitors (PMEIs). Int. J. Mol. Sci. 2018, 19, 2878. [Google Scholar] [CrossRef]
- Bolívar-Fernández, N.; Saucedo-Veloz, C.; Solís-Pereira, S.; Sauri-Duch, E. Maduración de frutos de saramuyo (Annona squamosa L.) desarrollados en Yucatán, México. Agrociencia 2009, 43, 133–141. [Google Scholar]
- Liu, W.; Zhang, J.; Jiao, C.; Yin, X.; Fei, Z.; Wu, Q.; Chen, K. Transcriptome analysis provides insights into the regulation of metabolic processes during postharvest cold storage of loquat (Eriobotrya japonica) fruit. Fruit Hortic. Res. 2019, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Weijin, L.; Tao, Z.; Jundi, Z.; Jinxiang, L.; Changchung, Y.; Kaidong, L. Comparative transcriptomic analysis of Split and non-split atemoya (Annona cherimola Mill. × Annona squamosa L.) fruit to identify potential genes involved in the fruit splitting process. Sci. Direct 2019, 248, 216–224. [Google Scholar] [CrossRef]
- Palomino-Hermosillo, Y.A.; Berumen-Varela, G.; Ochoa-Jiménez, V.A.; Balois-Morales, R.; Jiménez-Zurita, J.O.; Bautista-Rosales, P.U.; Martínez-González, M.E.; López-Guzmán, G.G.; Cortés-Cruz, M.A.; Guzmán, L.F. Transcriptome analysis of soursop (Annona muricata L.) fruit under postharvest storage identifies genes families involved in ripening. Plants 2022, 11, 1798. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Jiménez, V.A.; Balois-Morales, R.; López-Guzmán, G.G.; Jiménez-Zurita, J.O.; Felipe-Guzmán, L.; Berumen-Varela, G. Changes in quality and gene transcript levels of soursop (Annona muricata L.) fruits during ripening. Mex. J. Biotechnol. 2023, 8, 1–16. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A. IQ-TREE: A fast effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analysis of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Talavera, A.; Fernández-Pozo, N.; Matas, A.J.; Hormaza, J.I.; Bombarely, A. Genomics in neglected and underutilized fruits crops: A chromosome-scale genome sequence of cherimoya (Annona cherimola). Plants People Planet 2023, 5, 408–423. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleics Acids Res. 2008, 36, 3421–3435. [Google Scholar] [CrossRef]
- Louvet, R.; Cavel, E.; Gutiérrez, L.; Guénin, S.; Roge, D.; Gillet, F.; Guerineau, F.; Pelloux, J. Comprehensive expression profiling of the pectin methylesterase gene family during silique development in Arabidopsis thaliana. Planta 2006, 224, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, Y.; Wang, S.; Zhang, Q.; Yang, S. Genome-wide identification of PME genes, evolution and expression analyses in soybean (Glycine max L.). BMC Plant Biol. 2021, 21, 578. [Google Scholar] [CrossRef]
- Li, Z.X.; Wu, L.M.; Wang, C.; Wang, Y.; He, L.G.; Wang, Z.J.; Ma, X.F.; Bai, F.X.; Feng, G.Z.; Liu, J.H. Characterization of pectin methylesterase gene family and its possible role in juice sac granulation in navel orange (Citrus sinensis Obsbeck). BMC Genome 2022, 23, 185. [Google Scholar] [CrossRef]
- Tang, G.; Chen, G.; Ke, J.; Wang, J.; Zhang, D.; Liu, D.; Huang, J.; Zeng, S.; Liao, M.; Wei, X.; et al. The Annona montana genome reveals the development and flavor formation in mountain soursop fruit. Ornam. Plant Res. 2023, 3, 14. [Google Scholar] [CrossRef]
- Verma, C.; Mani-Tiwari, A.M.; Mishra, S. Biochemical and molecular characterization of cell wall degrading enzyme, pectin methylesterase versus banana ripening: An overview. Asian J. Biotechnol. 2017, 9, 1–23. [Google Scholar] [CrossRef]
- Müller, K.; Levesque, T.G.; Fernandes, A.; Wormit, A.; Bartels, S.; Usadel, B.; Kermode, A. Overexpression of a pectin methylesterase inhibitors in Arabidopsis thaliana leads to altered growth morphology of the stem and defective organ separation. Plant Signal. Behav. 2013, 8, e26464. [Google Scholar] [CrossRef]
- Zhu, X.; Tang, C.; Li, Q.H.; Qiao, X.; Li, X.; Cai, Y.L.; Wang, P.; Sun, Y.Y.; Zhang, H.; Zhang, S.L. Characterization of the pectin methylesterase inhibitor gene family in Rosaceae and role of PbrPMEI23/39/41 in methylesterified pectin distribution in pear pollen tube. Planta 2021, 253, 118. [Google Scholar] [CrossRef]
- Lionetti, V.; Raiola, A.; Mattei, B.; Bellincampi, D. The grapevine VvPMEI1 gene encodes a novel functional pectin methylesterase inhibitor associated to grape berry development. PLoS ONE 2015, 10, e0133810. [Google Scholar] [CrossRef]
- Martínez-González, M.E.; Balois-Morales, R.; Alia-Tejacal, I.; Cortes-Cruz, M.A.; Palomino-Hermosillo, Y.A.; López-Guzmán, G. Postharvest fruits: Maturation, softening and transcriptional control. Rev. Mex. Cienc. Agríc. 2017, 8, 4089–4101. [Google Scholar] [CrossRef][Green Version]
- Desphande, A.B.; Anamika, K.; Jha, V.; Chidley, H.G.; Oak, P.S.; Kadoo, N.Y.; Pujari, K.H.; Giri, A.P.; Gupta, V.S. Transcriptional transitions in Alphonso mango (Mangifera indica L.) during fruit development and ripening explain its distinct aroma and shelf-life characteristics. Sci. Rep. 2017, 7, 8711. [Google Scholar] [CrossRef]
- Zhai, Z.; Feng, G.; Wang, Y.; Sun, Y.; Peng, X.; Xiao, Y.; Zhang, X.; Jiao, J.; Wang, W.; Du, B.; et al. Genome-wide identification of xyloglucan endotransglucosylasa/hydrolase (XHT) and polygalacturonase (PG) genes and characterization of their role in fruit softening of sweet cherry. Int. J. Mol. Sci. 2021, 22, 12331. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hou, Q.; Wang, L.; Zhang, T.; Zhao, W.; Yan, T.; Zhao, L.; Li, J.; Wan, X. Genome-wide identification and characterization of polygalacturonase gene family in maize (Zea mays L.). Int. J. Mol. Sci. 2021, 22, 10722. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yeats, T.H.; Uluisik, S.; Rose, J.K.; Seymour, G.B. Fruit softening: Revisiting the role of pectin. Trends Plant Sci. 2018, 23, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Zurita, J.O.; Balois-Morales, R.; López-Guzmán, G.G.; Palomino-Hermosillo, Y.A.; Bautista-Rosales, P.U. Guanay-1, Guanay-2 y Guanay-3: Nuevas variedades de guanábana para Nayarit. Rev. Mex. Cienc. Agríc. 2023, 14, 641–646. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).