3.1. Comparison of Porosity Investigation Methods
In
Figure 2, the characterization methods for the microstructures of the positive electrodes are summarized and sorted according to the required sample amount for an accurate measurement. Therein, the comparatively large sample volume for the different methods, except SEM, has the advantage of yielding representative results by analyzing a complete electrode sheet with a size of approximately 150 cm
2. The width of the bars represents the range of pore sizes that can be investigated with the corresponding technique. Most of them are physically limited (e.g., by the size of the N
2-molecule or the pressure range of MIP), while the upper and lower limits of the FIB/SEM tomography technique were determined by a combination of desired resolution, sample volume, and measurement time. He-pycnometry does not directly measure the pores within the electrode, but helium gas penetrates all accessible pores (>kinetic diameter of He), and by measuring
ρskel and comparing with
ρxtal, it allows the determination of the amount of closed pores. This method has no upper pore size limit.
As an example, for the investigated microstructural features in dependence of the respective methods,
Figure 3 shows an embedded cross-section of a positive electrode. The CBD (not visible) fills most of the voids between the AM particles. With the gauge, only the highest electrode thickness value can be measured, including the current collector.
The compaction of the electrode to 30% porosity resulted in the cracking of several AM secondary particles. This led to an increase in the inner surface area and created pores in the nanometer range, which can be measured by N
2-physisorption. Particle cracking at the surface and clogging of surface pores with CBD caused a densification of the electrode surface, which can be identified by SEM or quantified via CLSM. The closing of surface pores with CBD is also described by Haselrieder et al. and Günther et al. as an effect of high densification of the electrodes [
41,
42]. The closed pores inside the AM secondary particles led to a difference between
ρskel and
ρxtal that can be measured by He-pycnometry. Most of the accessible pores within the composite electrode were in the measurable range of MIP. To verify all of the described microstructural features and effects, FIB/SEM tomography was necessary to create a 3D model of the electrode.
As a reference measurement for all further methods, the thicknesses of the electrodes determined with a gauge were used to calculate the total porosity. The uncalendered electrodes had a porosity of 44%. After calendering, porosities of 39%, 30%, 26%, and 18% were produced. To verify the reproducibility of the results, an electrode sheet, which was identically processed as the one with 30% porosity, was additionally considered. The total porosity of electrodes from this sheet was 28% (gauge measurement). The corresponding electrode densities, ρbulk, ranged from 2.47 g cm−3 (uncalendered) to 3.65 g cm−3 (18% porosity). Measuring 60 electrodes each, the standard deviation (=statistic error) was the largest for the electrode with the lowest porosity (18 ± 1.6%) and the smallest for the uncalendered electrode (44 ± 0.7%). This finding was attributed to the larger absolute influence of the inaccuracy of the gauge (±1 µm) for the thinner electrodes. Therefore, the systematic error of the measured porosity originating from the gauge was only ±0.6% for the electrode with the highest porosity and with an electrode thickness of 94 ± 1 µm but ±1.3% for the electrode with the lowest porosity (64 ± 1 µm). Considering the natural thickness inhomogeneity of the electrodes and the systematic error of the thickness measurement, the according overall standard deviation amounted to 0.7–1.6% porosity.
Consequently, the porosity values are specified as integer values, as accuracy to the decimal place cannot be reasonably accomplished for composite electrodes. This is important when comparing the results and errors of the different methods in this study and to values in the literature. A difference of 1–2% in porosity cannot be considered significant for most analytical methods. Therefore, the difference between the two identically processed electrodes with 30% and 28% porosity can be considered negligible as the deviations were within the margin of error.
3.3. He-Pycnometry
To measure the skeletal density,
ρskel, of the electrodes and the AM powder, He-pycnometry measurements were performed. By comparing
ρskel with
ρxtal, the closed porosity
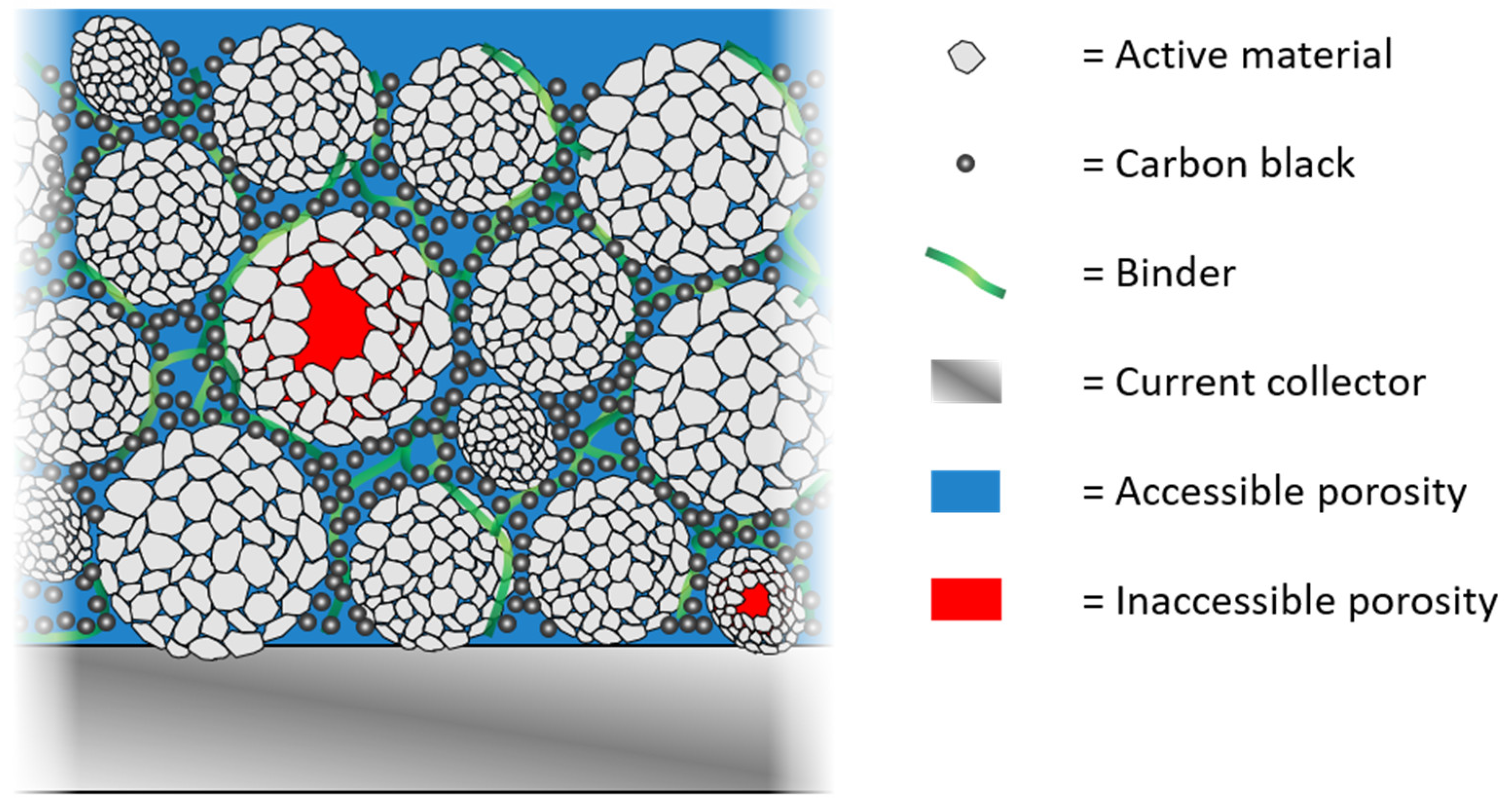
εinacc could be determined. Closed pores, as shown in
Figure 5(c2), can be located in AM secondary particles, in CBD agglomerates, between CBD and AM particles, or between AM particles. The latter could occur especially after strong calendering in combination with particle cracking.
The results for
ρskel ranged between 4.27 g cm
−3 for the uncalendered electrode and 4.31 g cm
−3 for the lowest porosity electrode. As the uncertainty was up to ±0.01 g cm
−3, the difference with regard to
ρskel for the variation in porosities can be considered to be insignificant. Comparing
ρskel with
ρxtal (4.436 g cm
−3) provides the amount of closed pores inside the solid volume of the electrode, which was between 3.7% and 2.8% (highest to lowest porosity). This amount agrees with the measurement of
ρskel (4.61 g cm
−3) and
ρxtal (4.76 g cm
−3) of the pristine AM powder. Here, the void space inside the AM particles was determined to be 3.2%. To some extent, the polycrystallinity of the AM also decreased its density in comparison to
ρxtal. The main contribution of the difference between
ρskel and
ρxtal of the AM remains the pores inside the AM particles as visualized in the SEM cross-sections (see
Figure 3). By He-pycnometry, the pores in the AM can be quantified.
The increase in ρskel with lower porosity could be due to the particle cracking of the AM. With this, the He-gas obtains access to originally closed pores. A counteracting effect could be the creation of new closed pores due to the intense calendering, especially within the CBD. However, due to the high amount of particle cracking at low total porosity identified in the SEM images, this was assumed to be the dominant effect for changes in ρskel.
In contrast to calculating closed pores inside the solid volume, calculating εinacc also took the void volume inside the electrodes into consideration, which then resulted in closed porosities of 2.2%/2.1%/2.6%/2.3%/2.4% (highest to lowest total porosity ε). The highest value for εinacc was calculated for electrodes with ε = 30%. This was confirmed by reference electrodes with ε = 28% and εinacc = 2.6%. Considering the margin of error of ρskel, the uncertainty for εinacc was 0.2 percentage points. Thus, the difference in εinacc among the different porous electrodes was not significant.
As a consequence of the calculation of εinacc, the accessible porosity, εacc, was for all electrodes more than two percentage points lower than ε, which affected the pore volume in the electrode that could be filled by electrolyte.
In addition to the rational results for
ρskel, errors in the measurement technique should be considered. The measured sample volume increased during the first two measurement steps (procedure described in
Section 2) and stabilized afterwards. Nguyen et al. studied ideal measurement conditions for different material classes (e.g., nonporous silicon and porous zeolite material) and the influence of activation (=drying) of the samples, the number of cycles, and percentage fill volumes (sample volume compared to volume of the measuring chamber) [
40]. Since the electrodes in our study were dried during sample preparation for N
2-physisorption measurements, the amount of residual moisture within the porous electrode structure was expected to be negligible. Before starting the pycnometry measurement, the samples were briefly exposed to the lab atmosphere during sample transfer (sample preparation in a dry room), possibly causing deviations during the first measurement cycles. The small kinetic diameter of He of 0.26 nm enables diffusion of the gas into the different electrode materials, especially into the CBD [
40]. However, changing to a larger or even non-inert gas molecule, such as N
2, would result in larger errors. The annulus volume, which is the volume between the surface of the electrode materials and the closest distance at which the gas molecules approach it, would increase and the ad-/desorption of N
2 by the sample would result in incorrect pressure readings [
40]. Another source of inaccuracy is the subtraction of the volume of the aluminum current collector, which was determined in a separate measurement. This additional measurement was performed with pristine foil.
The pycnometry measurement was necessary to calculate εacc, which describes the maximum pore volume that can be filled with electrolytes. According to the results of this study, 2–3% less electrolyte was needed than expected from the total pore volume within the electrodes. In addition, the closed pores within the solid phase could influence the mechanical properties of the electrodes (e.g., volume expansion of AM during de-/lithiation), and measuring a variation of these pores for differently compressed electrodes could be a sign of structural changes such as particle cracking of the AM.
3.4. Mercury Intrusion Porosimetry and N2-Physisorption
The most common technique to investigate the porosity and especially the PSD of LIB electrodes is MIP. As shown in
Figure 2, MIP covers nearly all pore sizes, which occur in electrodes of LIBs besides micropores. The results of the intrusion experiments are shown in
Figure 5. The specific volume of mercury (Hg),
νHg, within the electrodes during in-/extrusion is depicted in
Figure 5a. From the intrusion data, the PSD (
Figure 5b) can be calculated, assuming Hg is a perfect non-wetting liquid with a constant contact angle to the electrode material surface. Releasing pressure after intrusion results in an extrusion of Hg from the electrode. This process is influenced by the shape and size of the pores. For uncalendered electrodes, nearly all the intruded Hg stays trapped in the electrode after pressure release, while only half of the Hg remains inside in case of electrodes with the lowest porosity (marked in
Figure 5a). One reason could be the change in pore shape from spherical (for uncalendered electrodes) to more conical-like pores (for lower porosities). Spherical pores have a larger ratio of pore body size to pore throat size, which increases the amount of Hg-entrapment [
43]. Rigby and Edler suggest further reasons for the Hg-entrapment being spatially extended structural heterogeneities, a snap-off in narrow pores located next to much larger pore elements (marked in
Figure 5(c5)), and the contact angle hysteresis [
43].
A change in pore shape, even at constant porosity, alters the electronic and ionic conductivity of battery electrodes [
34]. Thus, the pore shape directly influences the electrochemical performance of LIBs and electrodes. Furthermore, the surface area of pores increases when changing from spherical to more conical and slit-like pores. This is important for interface chemistry (electrolyte additive research) and for calculating the ionic conductivity of the electrolyte within the electrode (tortuosity, currently under investigation). Additional insights are expected when comparing MIP and N
2-physisorption, discussed hereafter.
More structural heterogeneities of uncalendered electrodes compared to calendered electrodes and, therefore, a higher amount of Hg-entrapment can be confirmed with the PSD results in
Figure 5b. The PSD graph shows the differential specific pore volume with respect to the logarithm of the pore diameter (
dvHg/dlogD) as a function of the pore size. A bimodal distribution can be identified for all investigated electrode porosities with a major fraction of pore sizes (highest peak) at 3 µm for uncalendered electrodes. This peak shifts to smaller pore sizes for lower electrode porosities down to nearly 400 nm for electrodes with 18% total porosity. A second peak at ≈100 nm stays almost constant for all investigated electrode porosities and can be attributed to pores within or between CBD clusters [
27]. At pore sizes below 100 nm, the specific pore volume increases for the lower porosity electrodes. The reasons for this increase are, on the one hand, the compression of larger pores, and on the other hand the creation of smaller pores resulting from AM particle cracking, starting here at 30% porosity towards lower porosities.
The peak shift with lower porosity results in a decrease in structural heterogeneities because the PSD becomes more uniform. Contrary to this, the increase in the number of pores below 100 nm for the electrode with the lower porosity and the overall densification of the electrode resulted in a more tortuous pore network and, thus, a lower effective ionic conductivity within the electrode/electrolyte assembly.
In addition to the PSD, the bulk sample volume,
Vbulk (Hg), can be measured after covering the electrodes with Hg and before applying a pressure for intrusion, since Hg is considered to be a perfect non-wetting liquid. With
Vbulk (Hg) and the sample mass,
msample,
ρbulk (Hg) can be calculated. Furthermore, the total amount of intruded Hg
VHg yields information considering
εacc down to pore sizes of 3.7 nm. Reducing
Vbulk (Hg) by
VHg results in the apparent density
ρapp.
If the electrodes show no structural changes during MIP (e.g., no compression) and contain no pore throats with a size < 3.7 nm,
ρapp should be close to
ρskel (determined by He-pycnometry). By applying the different densities, various porosities can be calculated from the specific Hg-volume,
νHg, for example:
as the total porosity from MIP. Using
ρskel in Equation (3) yields the accessible porosity
εskel (Hg), where closed pores are excluded. With
ρapp, only the Hg-accessible (apparent) porosity
εapp (Hg) is considered as shown in
Figure 5(c4). The difference between
εskel (Hg) and
εapp (Hg) results in the amount of pore volume accessible for helium but not for mercury. Inaccessible regions for Hg could also be due to too small pore entries of <3.7 nm or due to pressure-induced changes in the microstructure, e.g., changes in the elastic CBD. All calculated porosities from MIP are summarized, in comparison to the total porosity
ε determined by gauge measurements, in
Table 2. In addition, the specific pore surface area
σpore was determined from PSD results.
The total porosity derived by gauge measurements tends to be higher than derived by MIP. The main reason is the inaccuracy of gauge measurements indicated in
Section 3.1. Electrodes with an
ε of 30% and the related reference electrodes with an
ε of 28% confirm this statement, since there is only an insignificant difference between the
εxtal (Hg) of these samples. Furthermore, a gauge measurement overestimates the thickness of an electrode since only the thickest position of the electrode determines the result. This effect is amplified by structural heterogeneities of the samples. Thus, the largest difference between
ε and
εxtal (Hg) could be identified for uncalendered electrodes. In summary, to compare electrode microstructures, the porosity derived by MIP should be preferred to the porosity derived by gauge measurements.
The difference between the accessible porosities εskel (Hg) and εapp (Hg) decreases from 2.5 (for ε = 44%) to 0.5 (for ε = 18%) percentage points. Since the pore shape changes with calendering, more pores are accessible for Hg-intrusion at lower electrode porosities. Another explanation is that the strong calendered electrodes are less compressible and, thus, more stable against the high intrusion pressure, which results in a measured porosity closer to the true electrode porosity. For applications, such as LIBs, εapp (Hg) could have more relevance in terms of electrochemical performance than εskel (Hg) since pore spaces with small entries of <3.7 nm, still accessible by He-gas, could be electrochemically inactive or cause high overpotentials due to the hindered Li-ion kinetics, especially at higher applied current densities. Further insights into microporosity could be gained by N2-physisorption.
As a result of
Figure 5, the change in pore shape and the increase in the pores with a size < 100 nm for lower electrode porosity should lead to a change in the specific pore surface area,
σpore. In
Table 2, an increase in
σpore from 1.3 (
ε = 44%) to 2.1 m
2 g
−1 (
ε = 18%) can be identified, thus confirming the previous findings. Although the pore structure of a composite electrode is usually a mixture of different pore shapes, a conical pore shape model was applied to calculate
σpore for all electrode porosities, because this shape allows the best description of the hysteresis of the Hg-intrusion and extrusion data in
Figure 5a. Measurement of the specific surface area independent of the pore shape can be achieved by N
2-physisorption, which can be incorporated into the evaluation of the strengths and limitations of the chosen pore shape model in the MIP results.
Despite the detailed insights into the electrode microstructure and porosity from MIP experiments, a correct interpretation of the results and deviations is crucial. The measurement principle of MIP, where Hg in the capillary forms together with the metal sleeve around the dilatometer a plate capacitor and the intruded Hg-volume is proportional to the change in capacitance, is accurate up to ±0.05 mm3. But further variance regarding VHg, the filling volume of Hg in the dilatometer, and changes in temperature can result in a realistic accuracy of ±2 mm3 for the determined porosity and up to ±5 mm3 for the density calculation (values given by device distributor). With VHg ranging between 87 and 260 mm3 for lowest and highest electrode porosity, respectively, a relative error of 1–6% has to be considered. As for all analytical techniques in this study, the sample preparation can influence the accuracy of the results (e.g., cutting the electrode sheet into samples).
In addition, the use of an aluminum foil as a typical cathode electrode current collector [
44] can lead to additional inaccuracies since Al and Hg can form an alloy [
45]. However, this barely influences the results as the native oxide layer of Al prevents a reaction with Hg before and during the MIP measurement [
45], which was confirmed by a baseline measurement of the pristine aluminum current collector. With complex systems, such as composite electrodes, a constant contact angle of 140° between Hg and the electrode materials is considered as approximation.
When comparing the PSD from MIP with results from other methods (e.g., SEM or N
2-physisorption) the definition of the pore size has to be considered, since MIP measures the pore entries that are of the same size as the pore bodies (e.g., cylindrical pores) or smaller (e.g., spherical pores). Furthermore, the speed of the pressure increase/decrease can shift the pore size determination (faster = shift to smaller pore sizes). Structural heterogeneities (see Hg-entrapment) and the densification of the electrode surface, described by the CLSM results, amplifies the pore-blocking effect and shifts the PSD results to smaller pore sizes [
46].
Since nearly all described effects influencing the results of MIP are similar for electrodes with the same composition, the comparison of different electrode porosities is reasonable. Furthermore, by comprehending the factors that influence PSD changes and comparing the results of MIP with other methods, such as SEM cross-sections, changes in the electrode manufacturing process can also be understood and analyzed.
Overall, MIP measurements have a higher accuracy and reproducibility for porosity determination than gauge measurements. In addition, insights into different porosities (total, accessible), pore shape, structural homogeneity, and specific pore surface area can be gained and result in a comprehensive overview of the electrode microstructure. Measuring the pore throats instead of pore bodies with MIP can be beneficial to understand ionic transport within an electrolyte, as ion mobility is limited by the narrowest passage on a pathway through the electrode pore network. Therefore, MIP is considered to be a major method for characterizing LIB composite electrodes.
Considering N2-physisorption, information about micro- (<2 nm), meso- (2–50 nm), small macropores (>50 nm) and the BET specific surface area, σBET, can be gained, which can support the correct interpretation of the MIP results.
The linear isotherms of the N
2-adsorption experiments are shown in
Figure 6a (type II isotherm [
14]). As determined by MIP, the specific inner surface area of the electrodes increased for lower porosity, which was attributed to cracking of secondary AM particles during calendering. Fitting of the linear region of the isotherms with the BET model resulted in a linear trend with regard to increasing
σBET at reduced total electrode porosities as shown in
Figure 6b. The specific surface area doubles from uncalendered electrodes (44%) to highly densified electrodes (18% porosity), which can be used as indicators for the quantification of the extent of particle cracking as a consequence of extensive calendering.
Due to the presence of micropores, the quantitative validity of the BET model is limited, as the processes of micropore filling and monolayer-multilayer adsorption might interfere [
14,
47]. Therefore, the results of the BET model represent an apparent specific surface area. From the
t-plot (
Figure S2), a micropore surface area of ≈0.3 m
2 g
−1 was found for all investigated electrode porosities. With a constant micropore surface area, the comparison of the BET surface areas remains valid. In comparison to the specific pore surface area from MIP, MIP showed approximately twice the values of N
2-physisorption (
Table 2 compared to
Figure 6b). One reason is the possible shift in the PSD to smaller values in the MIP measurement, which results in a higher amount of smaller pores and a higher surface area. Another reason lies in the use of different models of the two methods to calculate the specific surface area. The BET model based on the specific monolayer capacity of the adsorbent in the electrode and
σpore from MIP simplifies the pore network to geometric shapes (here: conical). The latter model was also affected by changes in the pore shape due to the calendering of the electrodes. Therefore,
σBET calculated from N
2-physisorption should be used to describe the structure of the porous electrodes and to calculate the tortuosity (quadratic correlation between surface area and tortuosity) based on the model of Carniglia [
16].
A change in the specific surface area can have a significant effect on the LIB performance and cycle life [
48]. Cracking of AM particles results in an increased active specific surface area, which, however, can ultimately lead to enhanced detrimental side reactions between the AM surface and electrolyte [
49].
Looking at the hysteresis of the isotherm (
Figure 6c, inserted graph, type H3 [
14]), mesopores were present in the calendered electrode, while they were absent in the uncalendered electrode showing no AM particle cracking or densification of the CBD. The shape of the hysteresis indicates the presence of slit-shaped pores, since the desorption branch was shallow and parallel to the adsorption branch over a wide pressure range [
50]. As with the MIP, the PSD showed an increasing pore volume below a 100 nm pore size for low porosity electrodes. The shape of the PSD curves was comparable to the MIP experiments in the respective pore size range (
Figure 6c; MIP shown as dotted line for 18% porosity). Therefore, it can be concluded that both methods are suitable for the analysis of LIB electrodes. At a pore size of 100 nm, when the BJH model reaches its limit of validity, a slightly larger pore volume for MIP can be identified. The desorption branch of the isotherm mostly describes the size of pore throats, such as those in MIP, but N
2-physisorption is less affected by pore shielding or comparable errors. In this case, the higher pore volume measured with MIP at pore sizes of approximately 100 nm could be explained by a shift in the PSD to smaller values, since larger pores are blocked from the intruding Hg by a barrier of pores with smaller pore diameters ≈ 100 nm as shown in
Figure 5c.
Quantifying the micropore volume is not possible with the BJH model but with a discrete Fourier transform calculation [
14]. However, due to the high complexity of the pore network and its heterogeneity, there is no simple model to describe the whole pore structure ranging from micro- to macropores with different pore shapes.
Considering the real electrode pore system in an LIB filled with electrolytes, micropore volume may not be crucial for a good electrochemical performance. Markoulidis et al. showed the influence of micropores on the performance of electrochemical double-layer capacitors [
51]. Therein, it was stated that below a 1.8 nm pore size, the solvated Li-ions start to change their coordination in a standard electrolyte (1 M LiPF6 in ethylene carbonate (EC) and ethyl methyl carbonate (EMC), EC:EMC 50:50 wt%). In addition, PF
6 ions were desolvated in micropores smaller than 1.4 nm and kinetics for the ion transport slows down. Thus, especially for fast charging/discharging in LIBs, micropores could be inactive in terms of Li-ion transportation. Considering other battery systems, such as solid-state batteries, micropores and small mesopores can have a detrimental effect, because they hinder Li-ion mobility due to the nonconductive gaps between the solid electrolyte and/or AM particles.
The accuracy of N
2-physisorption was illustrated by the electrode with 30% total porosity and the reference electrode (
ε = 28%). No variance between the two porosities could be identified for the linear isotherm and the PSD. The difference in the two
σBET was 2%. Considering the systematic error of the device (e.g., the accuracy of pressure reading) and the limitations of the experimental setup (e.g., a limited sample space in tube), a realistic inaccuracy for the adsorbed quantity and the surface area was up to 5–10%. Therefore, the deviation for the BET results (
ε = 30% compared to
ε = 28%) was negligible and the micropore volume was in the range of the variance of the pressure reading. For samples with a small specific surface area, changing the analysis gas to krypton could lead to more accurate results due to the higher sensitivity [
14]. To keep the electrode structure unchanged during the sample preparation, the degassing temperature was reduced to 120 °C compared to inorganic powder samples (approximately 200 °C) but remaining contaminations are possible, e.g., clogged micropores.
Despite these limitations, when measuring electrodes with a small specific surface area compared to powder samples, N2-physisorption provides a more accurate result for a specific surface area than MIP measurements and confirms the PSD results of MIP in the pore size range of mesopores and small macropores (<100 nm). By comparing these two methods, a correct interpretation of the MIP outcome is possible considering measurement artifacts such as pore shielding. The accuracy of both methods depends on the porosity of the investigated electrodes, with low porosity favoring N2-physisorption (especially for determination of σBET) and high porosity favoring MIP (especially determination of the pore volume). However, despite the drawbacks, a comprehensive overview of the electrode microstructure can only be gained by MIP measurements.
3.5. Focused Ion Beam/Scanning Electron Microscopy Tomography
The results of the previous methods can be verified using FIB/SEM tomography combined with a digital 3D model applying the GeoDict2020 software to gain further insights into the electrode microstructure. For this purpose, the electrode with the lowest porosity (18%) was investigated. Highly densified electrodes are rarely investigated in the literature, even though the low porosities are required to achieve high energy densities of the battery cell. Intense calendering results in extensive changes to AM particles in terms of cracking and distortion, whereby a 3D instead of 2D visualization is beneficial for a comprehensive understanding of microstructural properties.
Figure 7 shows a FIB-prepared cross-section with its microstructural features. It verifies the results regarding the presence of closed pores in AM particles, AM particle cracking predominantly at the electrode surface, and that most of the space between the AM particles was filled with CBD. The cracking of large AM particles (e.g., at the left border of the FIB cross-section) could result in theoretical access (e.g., He-gas) to originally closed pores. Pore sizes from small mesopores inside the CBD to macropores in the lower parts of the electrode could be detected. The larger macropore depicted in
Figure 7 illustrates the limitations of the previous methods. With MIP, the actual macropore would be measured at smaller pore sizes due to the small pore entry. For N
2-physisorption, the pore size was above the detection limit, and in an embedded cross-section, the epoxy resin would reduce the contrast between CBD and pore.
To finalize the results of the previous methods and to create a 3D model using tomography in the next step, the focus of the FIB/SEM experiments was on the microstructure and porosity of the electrode. Therefore, the measurement parameters were optimized for the visualization of the AM, the AM particle cracking, and the closed pores within the AM. In this work, an FIB current of 2 nA was used for visualizing a larger tomography volume, and the CBD was simulated in the void space between AM particles for 3D reconstruction.
To resolve the CBD structure and the according mesopores, the required FIB preparation parameters led to a tradeoff between a longer measurement time and smaller tomography volume. Only using low FIB currents (e.g., 20 pA [
27]) for the cross-section preparation of CBD could preserve the according structure. Otherwise, melting effects occur and result in preparational structural changes such as clogging of pores within the CBD. Therefore, in literature FIB/SEM tomography is typically combined with simulations [
27,
52,
53].
Comparing the structure of the electrode in this study with the results in the literature, significantly higher electrode total porosities, where
ε was at least 31% [
36] and often higher [
27,
52,
53] than in this study (
ε = 18%), have to be considered. Stronger calendering as applied herein resulted in more AM particle cracking and in a different, more densified CBD structure. The conductive agent-to-binder ratio was set herein to 2:3, whereas in the literature, it was often 1:1, which changes the binder-to-surface area ratio and, therefore, the CBD structure.
Since there are also electrode materials behind the sectional plane visible in
Figure 7, an automatized discrimination of the phases (i.e., AM, CBD, pores) is challenging. To transfer the tomography data of the electrode to a 3D model, a high contrast for the microstructural features (AM, particle cracking, and closed pores) within the sectional plane was used (
Figure 8b).
A digital 3D model can be created based on the FIB-prepared cross-sectional micrographs applying the GeoDict2020 software. The results are shown in
Figure 8, with an example of an FIB-prepared cross-section and the same position in the digital model. The size of the extracted volume (
Figure 8a and
Figure S3) was 24.2 × 14.7 × 17.9 µm (width × height × depth) with a voxel edge length of 37.2 nm in each direction. The statistical digital twin enabled a better in-depth understanding of the microstructure and the simulation or calculation of the electrochemical and mechanical electrode properties.
The CBD was simulated on basis of the PSD from the MIP experiment (
ε = 18%) (see
Figure 5 and
Figure 8c) and the ratio of the volumetric amount of AM to CBD in the electrode recipe (see
Table 1).
As the binder also covers the AM in the real composite electrodes, the volume of the CBD in the model could be overestimated. Other research groups adjusted the CBD structure to fit Li-ion or electronic conductivity measurements [
36,
37,
53].
In the literature, the porosity of CBD varies from 47% (nanoporosity [
53]) via 50% [
36] to 58% [
27]. The porosity of the digitally created CBD in this study was 50.2% with a minimal pore diameter of 37.2 nm, limited by the voxel length. When simulating CBD, the distribution in the electrode could be random, contact, or surface oriented [
36], and the connections within the CBD in blocking, fully open, and partially open configurations for Li-ion diffusion [
37]. In this study, the distribution of CBD was controlled by decreasing the surface energy and considering an open configuration for the channels within the CBD.
Since simulating the CBD cannot be achieved by applying a singular correct model, the best model for the respective application has to be identified, fitting the measurements of the microstructure or the electrochemical experiments (herein MIP).
To compare the porosity and PSD within the digital model with the previous results, GeoDict can perform a simulated MIP or granulometry experiment. The processes of the MIP were simulated as in the experiment with Hg intruding from all sides except the electrode bottom, close to the non-porous current collector, or only from the surface of the electrode considering larger electrode areas as in commercial applications [
38,
39]. The simulated CBD determined the PSD (see
Figure 8c) in the significant range from 19 to 1000 nm. Larger pores between AM secondary particles that were not filled with CBD had only a minor influence on the results of the MIP simulation in this pore size region. Thus, the relevance of the CBD and of the correct CBD simulation increase with lower electrode porosity (here 18%).
Since a few larger pores were created by the simulation, differences in the results can be identified depending on how the PSD was determined. If Hg was only allowed to intrude from the sample surface, as in most of the experiments, the pore volume of larger pores (>1 µm) was filled through smaller pore entries (<1 µm) at the dense electrode surface as shown in
Figure 5c. If Hg could intrude from all sides except from the current collector side or if granulometry was used, the larger pores were also depicted by modeling of the PSD. The MIP simulation and MIP experiment both had a D50 of the pores in the range of 200–260 nm. Using granulometry instead of MIP resulted in a shift of the PSD to higher values. This suggests that the pores had a smaller entrance than diameter or even a complete pore shielding effect occurred. In the MIP experiments, a conical model of the pores was used to calculate the PSD. Regardless of the model, the observation considering larger pores isolated within the highly densified electrode, which are only measured at higher pressures in the MIP experiment, is confirmed in
Figure 7.
The digital model of the electrode showed a lower total porosity than in the gauge measurement (16.1% instead of 18%) and, therefore, resulted in a slightly higher electrode density of 3.72 g cm
−3 compared to 3.65 g cm
−3. In addition,
εinacc had a value of 0.5%, more than four times smaller in the model compared to the value determined with He-pycnometry. Therefore,
εacc was also slightly smaller compared to the MIP experiments. However, the localization, which was not possible to detect with He-pycnometry, and quantification of closed pores (marked green in
Figure 8a and
Figure S3) within the AM is an advantage of the 3D reconstruction, as it is important for simulation of ionic transport in AM secondary particles and their mechanical properties, which is often neglected in the literature [
54]. The closed pores were distributed within the AM secondary particles and not only in the particle center as assumed from the results of the embedded cross-sections.
One reason for the difference in total porosity of up to 2 percentage points is the lower limit of the pore size in the model. The batch size in the simulation was chosen to be the voxel length of 37.2 nm. Therefore, in the MIP simulation, pore throat diameters smaller than 18.6 nm were not covered, while the experiment covered pore throats down to 3.7 nm. For the strongest calendered electrode, 3.7% of εacc were in this range in the MIP experiment. Hence, 0.6 percentage points must be added to εacc from the simulation (now 16.2%) to be comparable to the experiment.
Since the depth of the prepared cross-section for FIB/SEM tomography was not covering the overall electrode thickness, the extracted electrode section for the simulation was from the top third (close to the surface) of the electrode. To some extent, more particle cracking was observed at this surface-near position compared to parts closer to the current collector. This resulted in a slightly denser electrode and a lower total porosity of the investigated electrode section. This effect could be amplified by a heterogenous distribution of the CBD within the electrode, as only the ratio of AM to CBD from the electrode recipe was applied in the model. Binder migration during the drying process or a segregation during calendering could lead to higher and lower amounts of CBD at some positions in the electrode [
42,
55]. Contradicting this, embedded SEM cross-sections in
Figure 4 and
Figure S1 suggest a homogeneous distribution of all components.
The major difference between the experiments and the digital model was in the amount of εinacc, which was more than four times smaller in the model. Here, the absolute accuracy and the made assumptions (e.g., ρtheo of AM is ρxtal) of all the experimental methods have to be considered, as they can lead to large relative errors for the small values of εinacc. A similar effect could originate from the comparatively small electrode, thus sample section investigated by FIB/SEM tomography. More particle cracking of secondary particles resulted in more access points to originally closed pores in AM in the model. As stated, the CBD model did not include nanoporosity with diameters smaller than 37.2 nm and no closed pores associated with the CBD.
For the segmentation of the FIB/SEM 3D image data, a non-local means filter was applied besides others, and the threshold for segmentation between active material and pore was chosen manually. These two factors might lead to small errors during segmentation, especially regarding small pores and cracks in the active material. It must be assumed that during the segmentation process, information regarding small pores and cracks in the active material are lost and, therefore, that the percentage of closed pores in the structure is underestimated.
Overall, the FIB/SEM tomography is a comprehensive complementary method to the previous methods that improves the understanding of the electrode microstructure and allows for the evaluation of the different porosity measurements. The modeling of the CBD in particular shows how important the correct representation of the inactive components is and where mistakes can be made when considering just porosity from experimental data.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}