
Auger- and X-ray Photoelectron Spectroscopy at Metallic Li Material: Chemical Shifts Related to Sample Preparation, Gas Atmosphere, and Ion and Electron Beam Effects

Abstract
:1. Introduction and Motivation
- Thin electrical non-conducting deposits (oxides, fluorides, carbonates, and implanted Ar) on surfaces with a high amount of elemental Li (e.g., Li foil, highly lithiated graphite, LiAl/LiAlZn anode material) showed characteristic XP peaks which shifted by around 3 eV to higher BE, irrespective of simple charging [24,31];
- Differently from conducting deposits (e.g., Au, Cu, adventitious carbon) which show the “normal” BE positions, artificial carbon (graphite powder) is not clearly well defined at Li-metal containing surfaces;
- Electron bombardment (AES) of Li carbonate (on Li foil) also leads to the decomposition of the carbonate to Li oxide. This effect has a critical dose. In the presence of adventitious carbon contamination, even metallic Li can be produced [28,30]. With only X-ray excitation (XPS), no significant damage can be observed at this point;
- Ar+ bombardement (depth profiling) of graphite that is not fully lithiated (HOPG) leads to the enrichment of Li at the surface. A partly carbide formation is assumed. After the measurements are electron-induced (AE spectra or mapping), out-diffusion (lateral and in-depth) of the Li from the measurement region is observed [28,29].
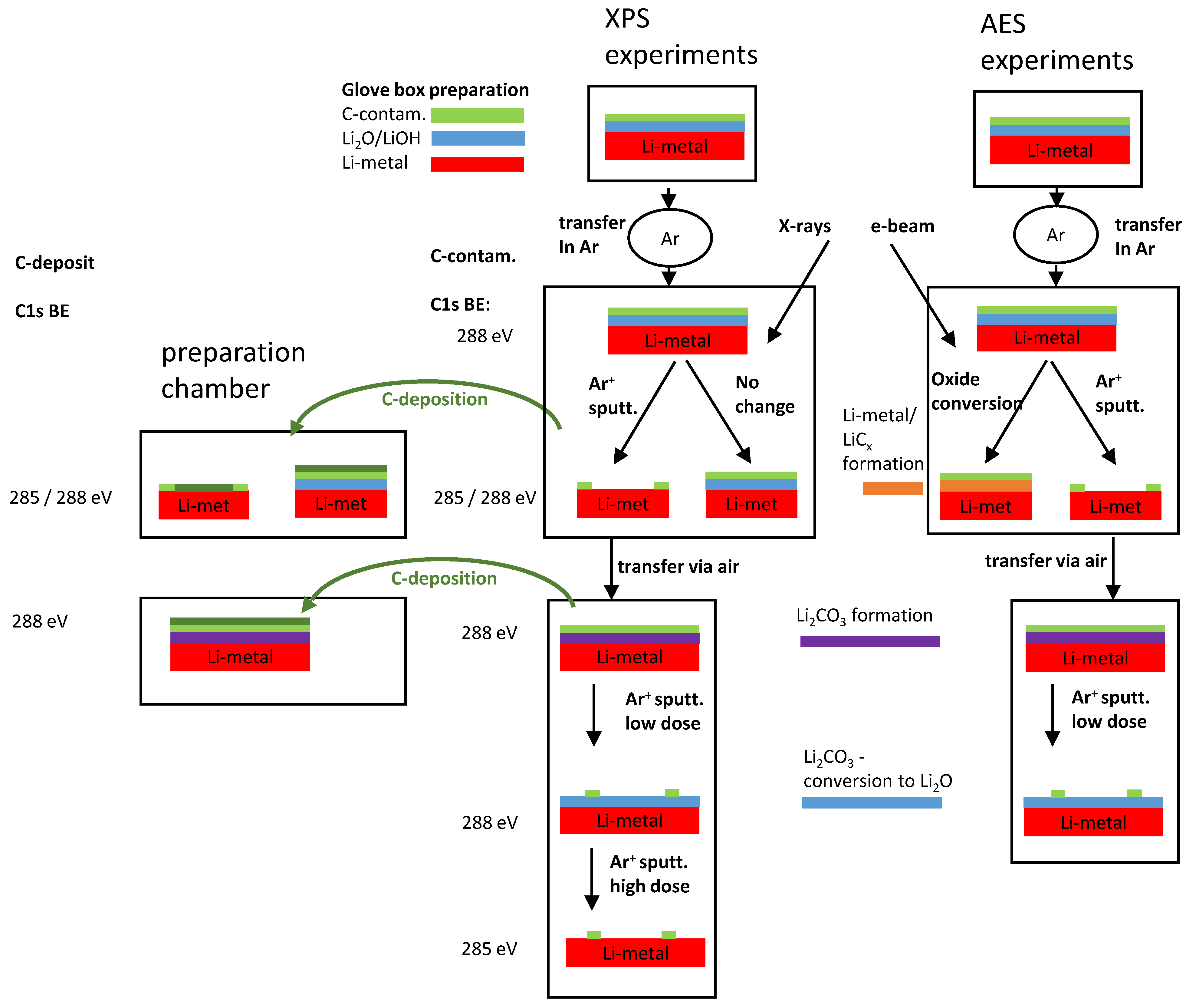
- Preparation of well-defined sample states comparable for both XP and AE analysis: We decided to use metallic Li as an experimental basis because it is the natural “key element” for materials used in all kinds of Li batteries. Because the previously used commercial Li foil was often contaminated with different species (mostly carbonate, partly oxy-fluorides, phosphates), we used Li massive material freshly cut in the glove box. Afterwards, the analysis chambers are transferred using an Ar filled transfer chamber. Careful analysis of the states is then performed, depending on vacuum and storage conditions. Metallic Li surfaces are then prepared with Ar+ sputtering, pure carbonate is formed with short exposition to air, and both XPS and AES are investigated at similar sample states.
- Introducing of new “internal standards” for BE referencing: Referencing carbon contamination is still the most common method. Because the first experiments with graphite powder as an artificial standard did not turn out to be unequivocal, we then tried to use (quasi) in situ thin film carbon deposition in preparation chambers coupled with the XPS apparatus. We used two different deposition methods: magnetron sputtering from a graphite target (only fine vacuum conditions) and thermal deposition of C60 (ultra-high vacuum conditions). Differently prepared surface states were considered for the C deposition experiments.
- Classification of e-beam and ion beam damage and its consequence for XP and AE analysis: In general, the damage by electron or ion radiation is well known, especially for oxide-type materials. Otherwise, such beams are commonly used for the excitation of analytical signals or for sample cleaning or element depth profiling. For the alkaline elements (as lithium), a high mobility is to be expected, even in the presence of near-surface electrical fields. Consequences for changes in the chemical states and the energy referencing are discussed for both XPS and AES, and area dependencies are also considered.
2. Experiments
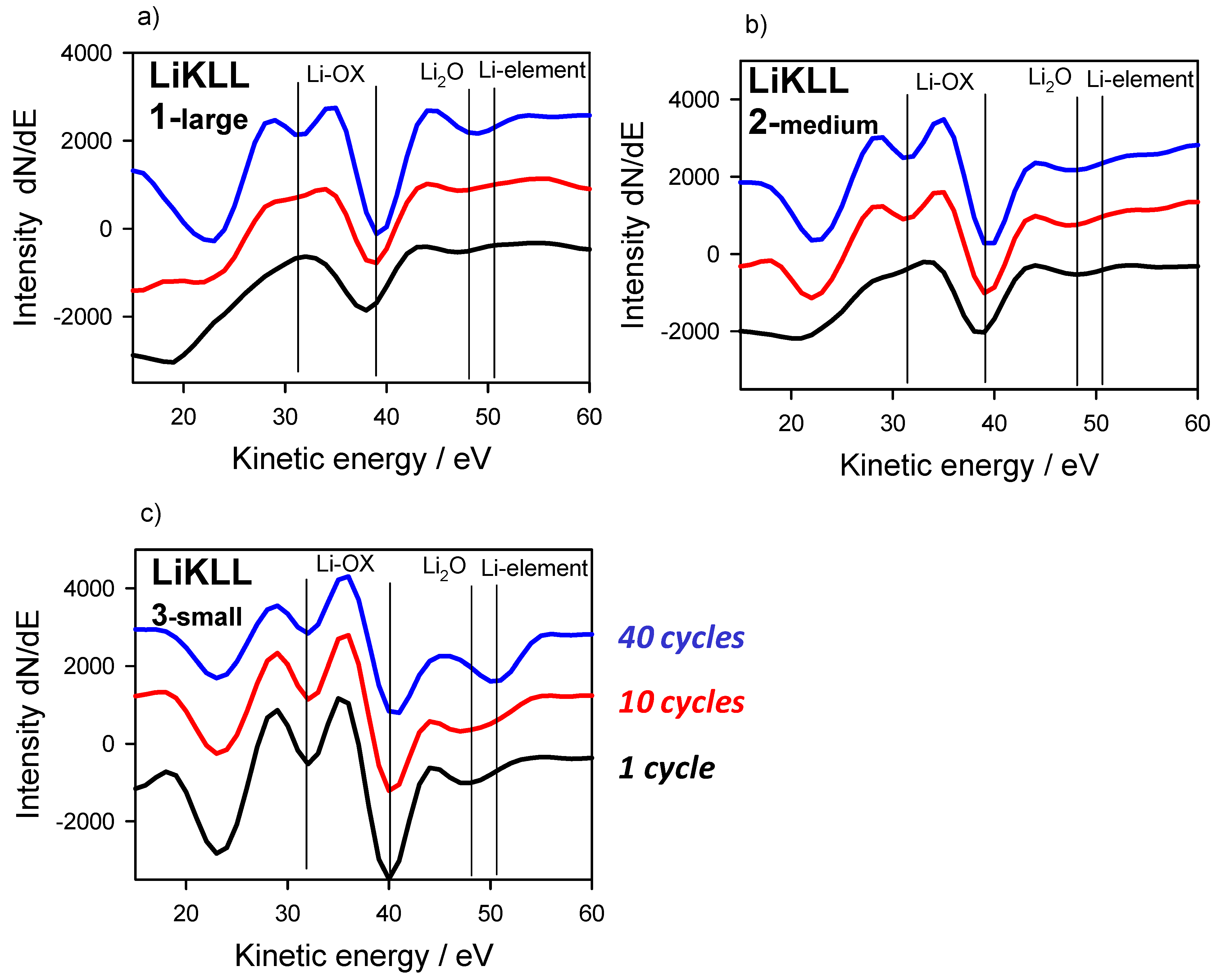
3. Results and Discussion
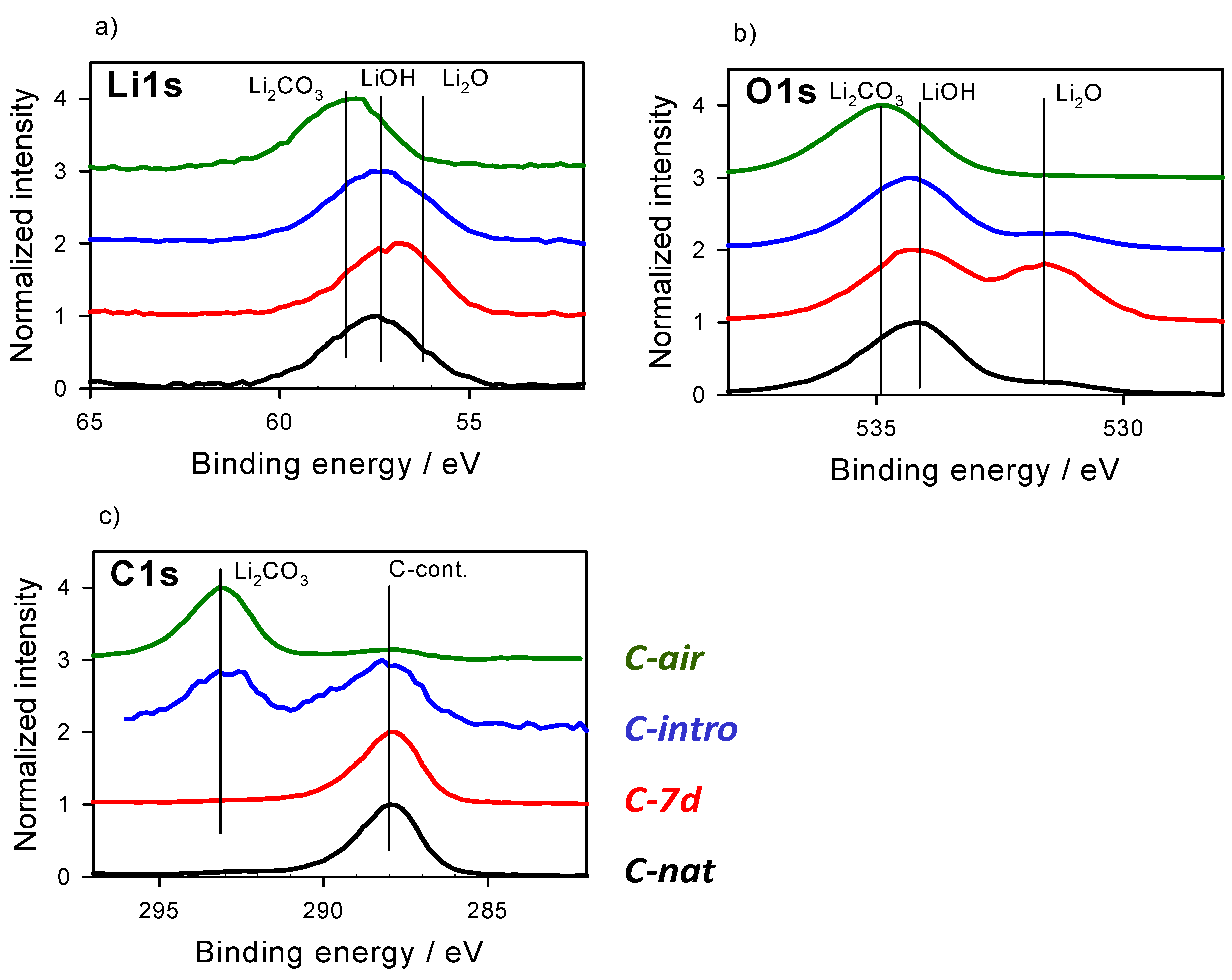
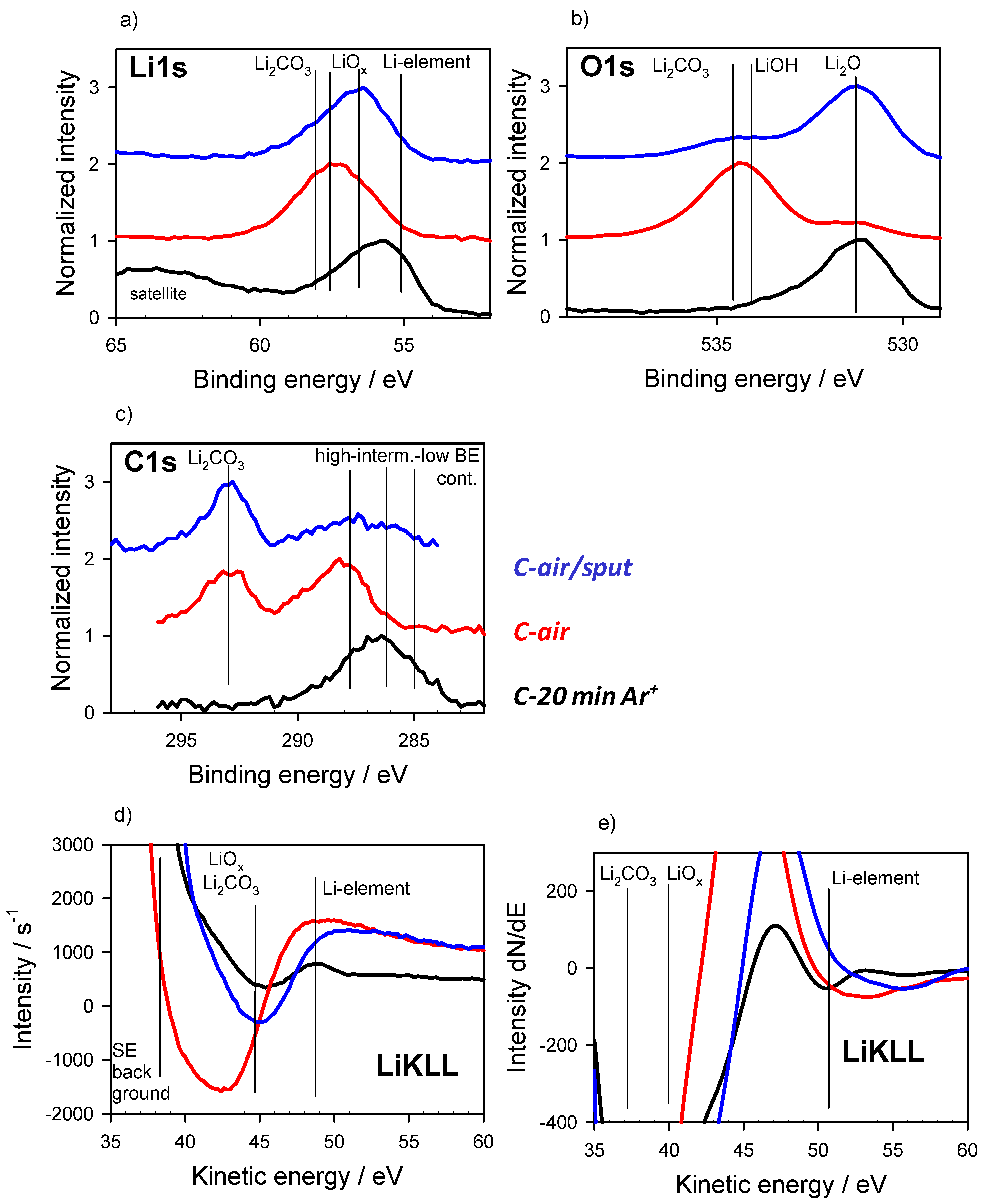
3.1. Chemical States as Prepared and with Gas Interaction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Li | O | C | Comment | |||||
|---|---|---|---|---|---|---|---|---|---|
| Li1s | Li1s | Li1s | O1s | O1s | O1s | C1s | C1s | ||
| Species | Li2CO3 | LiOH {Li2O2} | Li2O | Li2CO3 | LiOH {Li2O2} | Li2O | Li2CO3 | C-cont. | |
| Peak areas % | |||||||||
| on-air | 97 | 3 | 0 | 99 | <1 | <1 | 85 | 14 | |
| B-nat | <1 | 36 | 63 | 0 | 53 | 47 | 5 | 95 | |
| A-nat | 22 | 78 | 0 | 13 | 84 | 1 | 30 | 70 | |
| BE values | |||||||||
| Our fit energies | 58.0 | 57.5 | 56.4 | 534.6 | 534.1 | 531.3 | 292.8 | 288.2 | |
| Reference energies | 58.05 | 57.4 {57.5} | 56.4 | 534.67 | 533.77 {534.15} | 531.2 | 292.9 | 288.0 | Ref [27] |
| Reference energies | 55.5 | 54.8 | 53.7 | 532 | 531.0 | 528.5 * | 290.1 | 285.0 *** | Ref [36] |
| Difference to [27] | 2.5 | 2.7 | 2.7 | 2.6 | 3.1 ** | 2.7 | 2.7 | 3.2 *** |
| Conc.-Total | Li -Total | O -Total | C -Total | Comment | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conc.-fit | Li1s | Li2CO3 | LiOH | Li2O | O1s | Li2CO3 | LiOH | Li2O | C1s | Li2CO3 | C-cont. | |
| on-air | 36 | 36 | 0 | 0 | 49 | 49 | 0 | 0 | 15 | 14 | 1 | Li2CO3 |
| B-nat | 55 | 0 | 21 | 34 | 37 | 0 | 19 | 18 | 8 | 0.5 | 7 | LiOH + Li2O |
| A-nat | 45 | 9 | 36 | 0 | 48 | 6 | 42 | 0 | 7 | 3 | 4 | LiOH + (Li2CO3) |
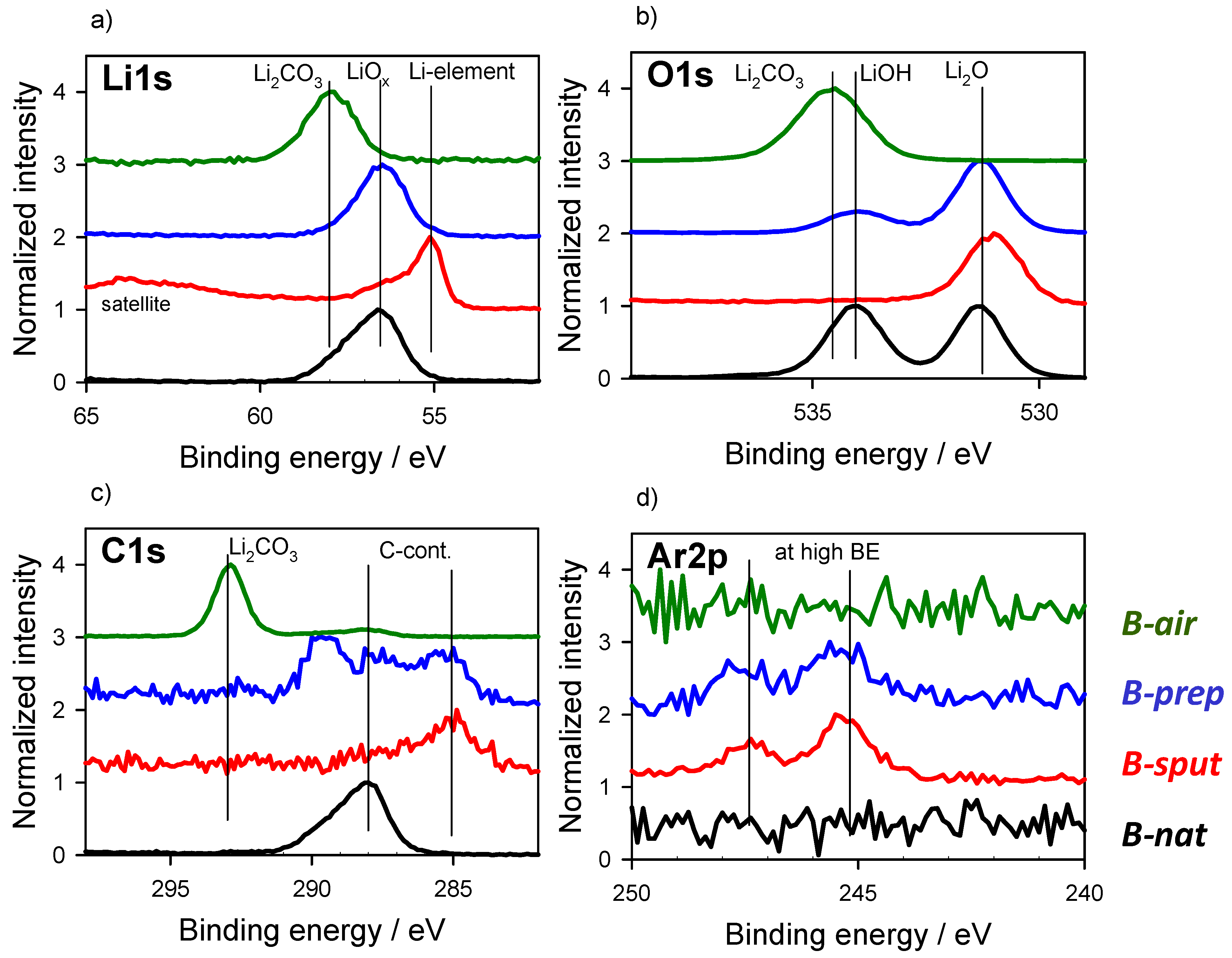
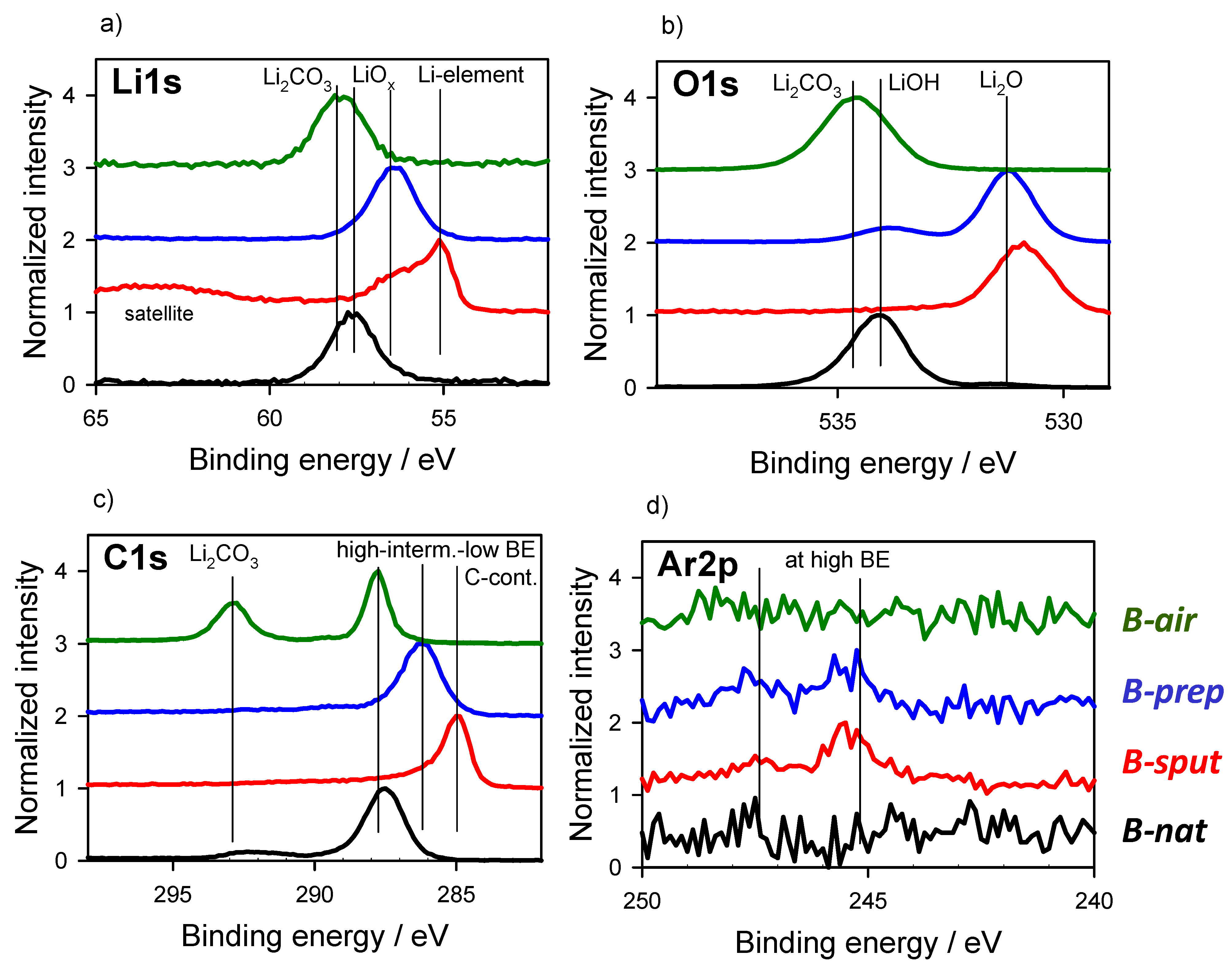
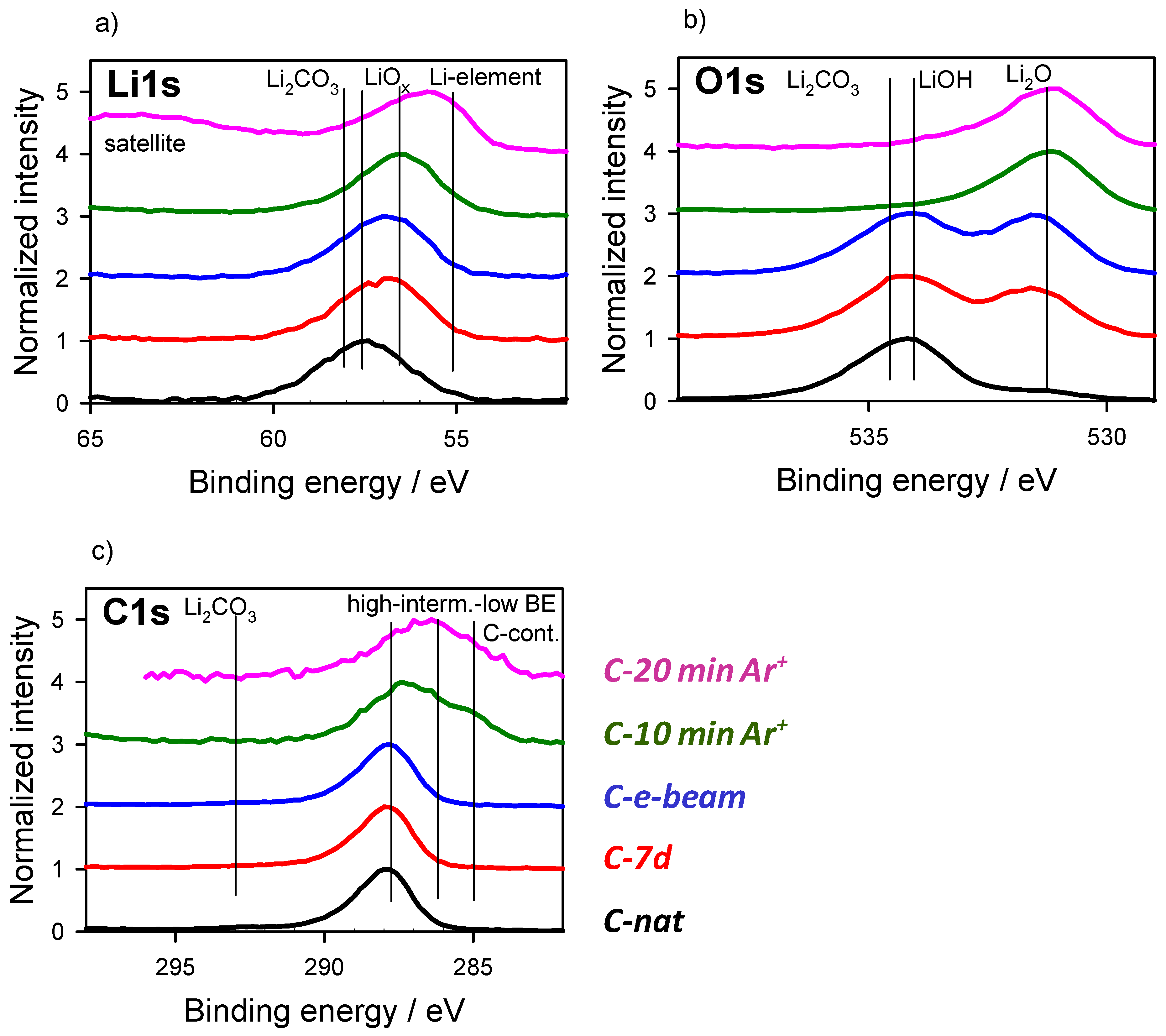
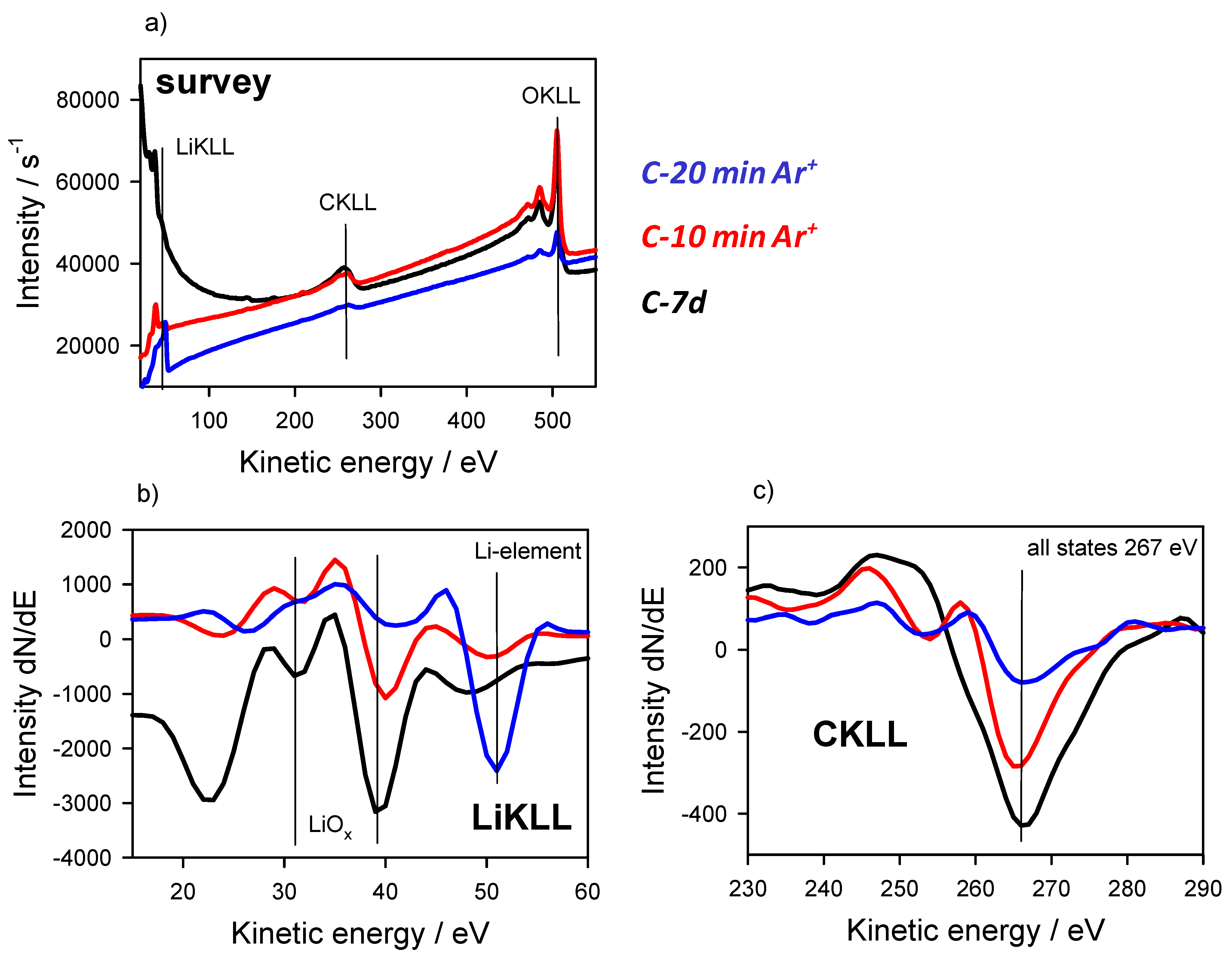
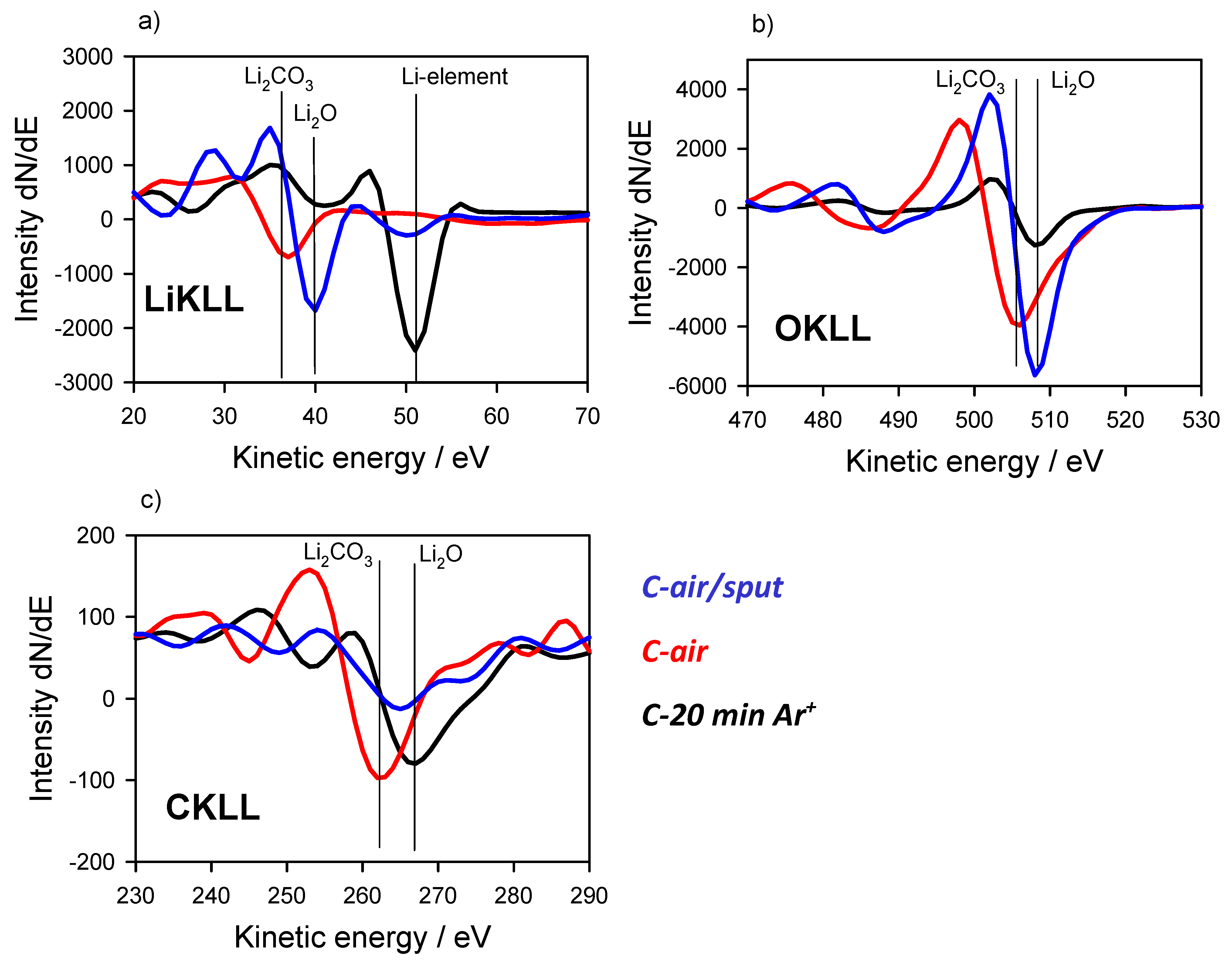
3.2. Energy Referencing for Sputter and Gas Modification: Artificial C and Ar
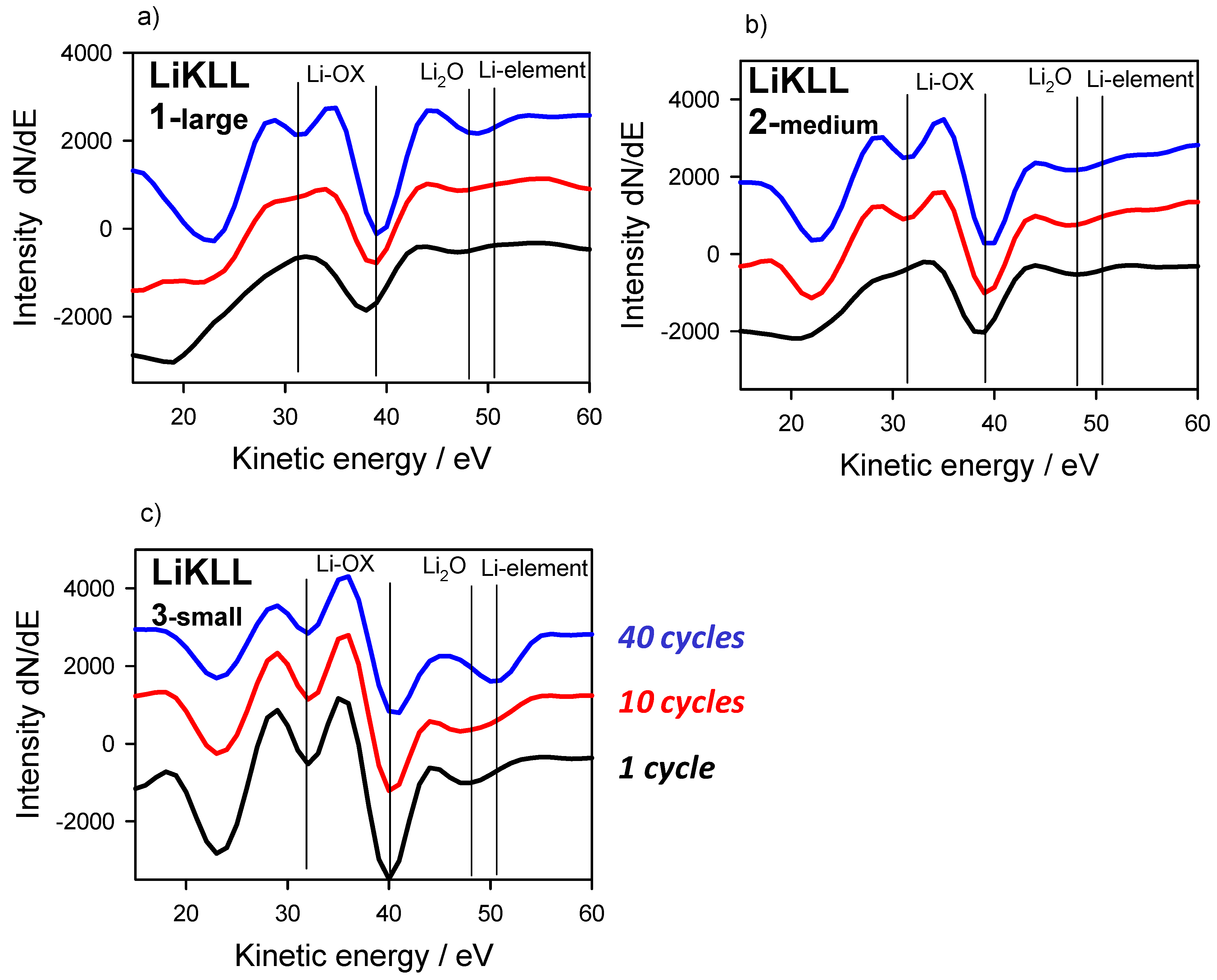
3.3. Large-Area Beam Modification in XP and AE Spectra
3.4. Area Dependent E-Beam Modification in AE Analysis
4. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Daniel, C.; Besenhard, J.O. (Eds.) Handbook of Battery Materials, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research development on sodium-ion batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Maire, P.; Novák, P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochim. Acta 2010, 55, 6332–6341. [Google Scholar] [CrossRef]
- Peled, E.; Menkin, S. Review—SEI: Past, present and future. J. Electrochem. Soc. 2017, 164, A1703–A1719. [Google Scholar] [CrossRef]
- Tang, C.-Y.; Leung, K.; Haasch, R.T.; Dillon, S.J. LiMn2O4 Surface Chemistry Evolution during Cycling Revealed by in Situ Auger Electron Spectroscopy and X-ray Photoelectron Spectroscopy. ACS Appl. Mater. Interfaces 2017, 9, 33968–33978. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.-Y.; Feng, L.; Haasch, R.T.; Dillon, S.J. Surface redox on Li[Ni1/3Mn1/3Co1/3]O2 characterized by in situ X-ray photoelectron spectroscopy and in situ Auger electron spectroscopy. Electrochim. Acta 2018, 277, 197–204. [Google Scholar] [CrossRef]
- Wood, N.K.; Steirer, K.X.; Hafner, S.E.; Ban, C.; Santhanagopalan, S.; Lee, S.-H.; Teeter, G. Operando X-ray photoelectron spectroscopy of solid electrolyte interphase formation and evolution in Li2S-P2S5 solid-state electrolytes. Nat. Comm. 2018, 9, 2490. [Google Scholar] [CrossRef]
- Abe, K.; Yoshitake, H.; Kitakur, T.; Hattori, T.; Wang, H.; Yoshio, M. Additives-containing functional electrolytes for suppressing electrolyte decomposition in lithium-ion batteries. Electrochim. Acta 2004, 49, 4613–4622. [Google Scholar] [CrossRef]
- Shima, M.; Tsutsumi, K.; Tanaka, A.; Onodera, H.; Tazawa, T. Quantitative Analysis of a Lithium Ion Battery Cathode Material with X-ray Photoelectron Spectroscopy and Auger Electron Spectroscopy. Microsc. Microanal. 2015, 21, 1356–1357. [Google Scholar] [CrossRef] [Green Version]
- Morigaki, K.-I.; Ohta, A. Analysis of the surface of lithium in organic electrolyte by atomic force microscopy, Fourier transform infrared spectroscopy and scanning Auger electron microscopy. J. Power Sources 1998, 76, 159–166. [Google Scholar] [CrossRef]
- Mori, S.; Asahina, H.; Suzuki, H.; Yonei, A.; Yokoto, K. Chemical properties of various organic electrolytes for lithium rechargeable batteries 1. Characterization of passivating layer formed on graphite in alkyl carbonate solutions. J. Power Sources 1997, 68, 59–64. [Google Scholar] [CrossRef]
- Kominato, A.; Yasukawa, E.; Sato, N.; Ijuuin, T.; Asahina, H.; Mori, S. Analysis of surface films on lithium in various organic electrolytes. J. Power Sources 1997, 68, 471–475. [Google Scholar] [CrossRef]
- Kalaga, K.; Shkro, I.A.; Haasch, R.T.; Peebles, C.; Bareño, J.; Abraham, D.P. Auger Electrons as Probes for Composite Micro- and Nanostructured Materials: Application to Solid Electrolyte Interphases in Graphite and Silicon-Graphite Electrodes. J. Phys. Chem. C 2017, 121, 23333–23346. [Google Scholar] [CrossRef]
- Pantano, C.G.; Madey, T.E. Electron beam damage in Auger electron spectroscopy. Appl. Surf. Sci. 1981, 7, 115–141. [Google Scholar] [CrossRef]
- Ohuchi, F.; Oginot, M.; Holloway, P.H.; Pantano, C.G., Jr. Electron Beam Effects During Analysis of Glass Thin Films with Auger Electron Spectroscopy. Surf. Interface Anal. 1980, 2, 85–90. [Google Scholar] [CrossRef]
- Ishida, N.; Fujita, D. Chemical-state imaging of Li using scanning Auger electron microscopy. J. Electron Spectrosc. Relat. Phenom. 2013, 186, 39–43. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, M.; Alvarado, J.; Wang, S.; Sina, M.; Lu, B.; Bouwer, J.; Xu, W.; Xiao, J.; Zhang, J.G.; et al. New Insights on the Structure of Electrochemically Deposited Lithium Metal and Its Solid Electrolyte Interphases via Cryogenic TEM. Nano Lett. 2017, 17, 7606–7612. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Pei, A.; Yan, K.; Sun, Y.; Wu, C.L.; Joubert, L.M.; Chin, R.; Koh, A.L.; Yu, Y.; et al. Atomic Structure of Sensitive. Battery Materials and Interfaces Revealed by Cryo−Electron Microscopy. Science 2017, 358, 506–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Z.; Wynn, T.A.; Schroeder, M.A.; Alvarado, J.; Wang, X.; Xu, K.; Meng, Y.S. Cryogenic Focused Ion Beam Characterization of Lithium Metal Anodes. ACS Energy Lett. 2019, 4, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Mitlin, D. Emerging Potassium Metal Anodes: Perspectives on Control of the Electrochemical Interfaces. Acc. Chem. Res. 2020, 53, 1161–1175. [Google Scholar] [CrossRef]
- Zheng, J.; Archer, L.A. Controlling electrochemical growth of metallic zinc electrodes: Toward affordable rechargeable energy storage systems. Sci. Adv. 2021, 7, eabe0219. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-C.; Gong, M.; Lu, B.; Wu, Y.; Wang, D.Y.; Guan, M.; Angell, M.; Chen, C.; Yang, J.; Hwang, B.-J.; et al. An ultrafast rechargeable aluminium-ion battery. Nature 2015, 520, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yuan, Y.; Liu, J.; Fichtner, M.; Pan, F. A review on current anode materials for rechargeable Mg batteries. J. Magnes. Alloys 2020, 8, 963–979. [Google Scholar] [CrossRef]
- Oswald, S. Binding energy referencing for XPS in alkali metal-based battery materials research (I): Basic model investigations. Appl. Surf. Sci. 2015, 351, 492–503. [Google Scholar] [CrossRef]
- Maibach, J.; Lindgren, F.; Eriksson, H.; Edström, K.; Hahlin, M. Electric Potential Gradient at the Buried Interface between Lithium-Ion Battery Electrodes and the SEI Observed Using Photoelectron Spectroscopy. J. Phys. Chem. Lett. 2016, 7, 1775–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, F.; Rehnlund, D.; Källquist, I.; Nyholm, L.; Edström, K.; Hahlin, M.; Maibach, J. Breaking Down a Complex System: Interpreting PES Peak Positions for Cycled Li-Ion Battery Electrodes. J. Phys. Chem. C 2017, 121, 27303–27312. [Google Scholar] [CrossRef]
- Wood, K.N.; Teeter, G. XPS on Li-Battery-Related Compounds: Analysis of Inorganic SEI Phases and a Methodology for Charge Correction. ACS Appl. Energy Mater. 2018, 1, 4493–4504. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M. Auger Elektronenspektroskopie an Lithium. Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany, 2019. [Google Scholar]
- Hoffmann, M.; Oswald, S.; Zier, M.; Eckert, J. Auger and X-ray photoelectron spectroscopy on lithiated HOPG. Surf. Interface Anal. 2016, 48, 501–504. [Google Scholar] [CrossRef]
- Hoffmann, M.; Zier, M.; Oswald, S.; Eckert, J. Challenges for lithium species identification in complementary Auger and X-ray photoelectron spectroscopy. J. Power Sources 2015, 288, 434–440. [Google Scholar] [CrossRef]
- Oswald, S.; Thoss, F.; Zier, M.; Hoffmann, M.; Jaumann, T.; Herklotz, M.; Nikolowski, K.; Scheiba, F.; Kohl, M.; Giebeler, L.; et al. Binding Energy Referencing for XPS in Alkali Metal-Based Battery Materials Research (II): Application to Complex Composite Electrodes. Batteries 2018, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Oswald, S.; Hoffmann, M.; Zier, M. Peak position differences observed during XPS sputter depth profiling of the SEI on lithiated and delithiated carbon-based anode material for Li-ion batteries. Appl. Surf. Sci. 2017, 401, 408–413. [Google Scholar] [CrossRef]
- Vogel, U.; Brachmann, E.; Oswald, S.; Menzel, S.; Gemming, T.; Eckert, J. Evaluation of a mobile vacuum transfer system for in vacuo XPS analysis using as-deposited Ti thin-films. Vacuum 2015, 117, 81–84. [Google Scholar] [CrossRef]
- Vogel, U. Grenzflächenausbildung zwischen LiNbO3 (LiTaO3) und Barriereschichten für den Einsatz bei Metallisierungssystemen für SAW-Strukturen. Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany, 2016. [Google Scholar]
- MultiPak, Software Package, V. 9.5; ULVAC-PHI: Osaka, Japan, 1994–2014.
- Kozen, A.C.; Pearse, A.J.; Lin, C.-F.; Schroeder, M.A.; Noked, M.; Lee, S.B.; Rubloff, G.W. Atomic layer deposition and in situ characterization of ultraclean lithium oxide and Lithium hydroxide. J. Phys. Chem. 2014, 118, 27749–27753. [Google Scholar] [CrossRef]
- Yao, K.P.C.; Kwabi, D.G.; Quinlan, R.A.; Mansour, A.N.; Grimaud, A.; Lee, Y.-L.; Lu, Y.-C.; Shao-Horn, Y. Thermal Stability of Li2O2 and Li2O for Li-Air Batteries: In Situ XRD and XPS Studies. J. Electrochem. Soc. 2013, 160, A824–A831. [Google Scholar] [CrossRef]
- Lesiak, B.; Mrozek, P.; Jablonski, A.; Jozwik, A. Analysis of the Auger KLL Spectra of Carbon by the Pattern Recognition Method. Surf. Interface Anal. 1986, 8, 121–126. [Google Scholar] [CrossRef]
- Smetyukhova, T.N.; Druzhinin, A.V.; Podgorny, D.A. Features of the Auger Spectra of Ti2C, SiC, and WC. J. Surf. Investig. X-ray Synchrotron Neutron Tech. 2017, 11, 414–419. [Google Scholar] [CrossRef]
- Bondarchuk, O.; LaGrow, A.P.; Kvasha, A.; Thieu, T.; Ayerbe, E.; Urdampilleta, I. On the X-ray photoelectron spectroscopy analysis of LiNixMnyCozO2 material and electrodes. Appl. Surf. Sci. 2021, 535, 147699. [Google Scholar] [CrossRef]
- Azmi, R.; Trouillet, V.; Strafela, M.; Ulrich, S.; Ehrenberg, H.; Bruns, M. Surface analytical approaches to reliably characterize lithium ion battery electrodes. Surf. Interface Anal. 2018, 50, 43–51. [Google Scholar] [CrossRef]
- Greczynski, G.; Hultman, L. X-ray photoelectron spectroscopy: Towards reliable binding energy referencing. Prog. Mater. Sci. 2020, 107, 100591. [Google Scholar] [CrossRef]
- Cheng, X.B.; Zhang, R.; Zhao, C.-Z.; Zhang, Q. Toward Safe Lithium Metal Anode in Rechargeable Batteries: A Review. Chem. Rev. 2017, 117, 10403–10473. [Google Scholar] [CrossRef]
- Maibach, J.; Källquist, I.; Andersson, M.; Urpelainen, S.; Edström, K.; Rensmo, H.; Siegbahn, H.; Hahlin, M. Probing a battery electrolyte drop with ambient pressure photoelectron spectroscopy. Nat. Commun. 2019, 10, 3080. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Kadam, S.; Li, H.; Shi, S.; Qi, Y. Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries. NPJ Comput. Mater. 2018, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Menkin, S.; O’Keefe, C.A.; Gunnarsdóttir, A.B.; Dey, S.; Pesci, F.M.; Shen, Z.; Aguadero, A.; Grey, C.P. Toward an Understanding of SEI Formation and Lithium Plating on Copper in Anode-Free Batteries. J. Phys. Chem. C 2021, 125, 16719–16732. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oswald, S. Auger- and X-ray Photoelectron Spectroscopy at Metallic Li Material: Chemical Shifts Related to Sample Preparation, Gas Atmosphere, and Ion and Electron Beam Effects. Batteries 2022, 8, 24. https://doi.org/10.3390/batteries8030024
Oswald S. Auger- and X-ray Photoelectron Spectroscopy at Metallic Li Material: Chemical Shifts Related to Sample Preparation, Gas Atmosphere, and Ion and Electron Beam Effects. Batteries. 2022; 8(3):24. https://doi.org/10.3390/batteries8030024
Chicago/Turabian StyleOswald, Steffen. 2022. "Auger- and X-ray Photoelectron Spectroscopy at Metallic Li Material: Chemical Shifts Related to Sample Preparation, Gas Atmosphere, and Ion and Electron Beam Effects" Batteries 8, no. 3: 24. https://doi.org/10.3390/batteries8030024
APA StyleOswald, S. (2022). Auger- and X-ray Photoelectron Spectroscopy at Metallic Li Material: Chemical Shifts Related to Sample Preparation, Gas Atmosphere, and Ion and Electron Beam Effects. Batteries, 8(3), 24. https://doi.org/10.3390/batteries8030024