
Enhancing CO2 Adsorption on MgO: Insights into Dopant Selection and Mechanistic Pathways


Abstract

1. Introduction
2. Materials and Methods
2.1. Theoretical Simulation of CO2 Adsorption
2.2. Materials
2.3. Preparation of MgO and C-Doped MgO Nanoparticles
2.4. Morphology Characterization and CO2 Adsorption Measurement
3. Results and Discussion
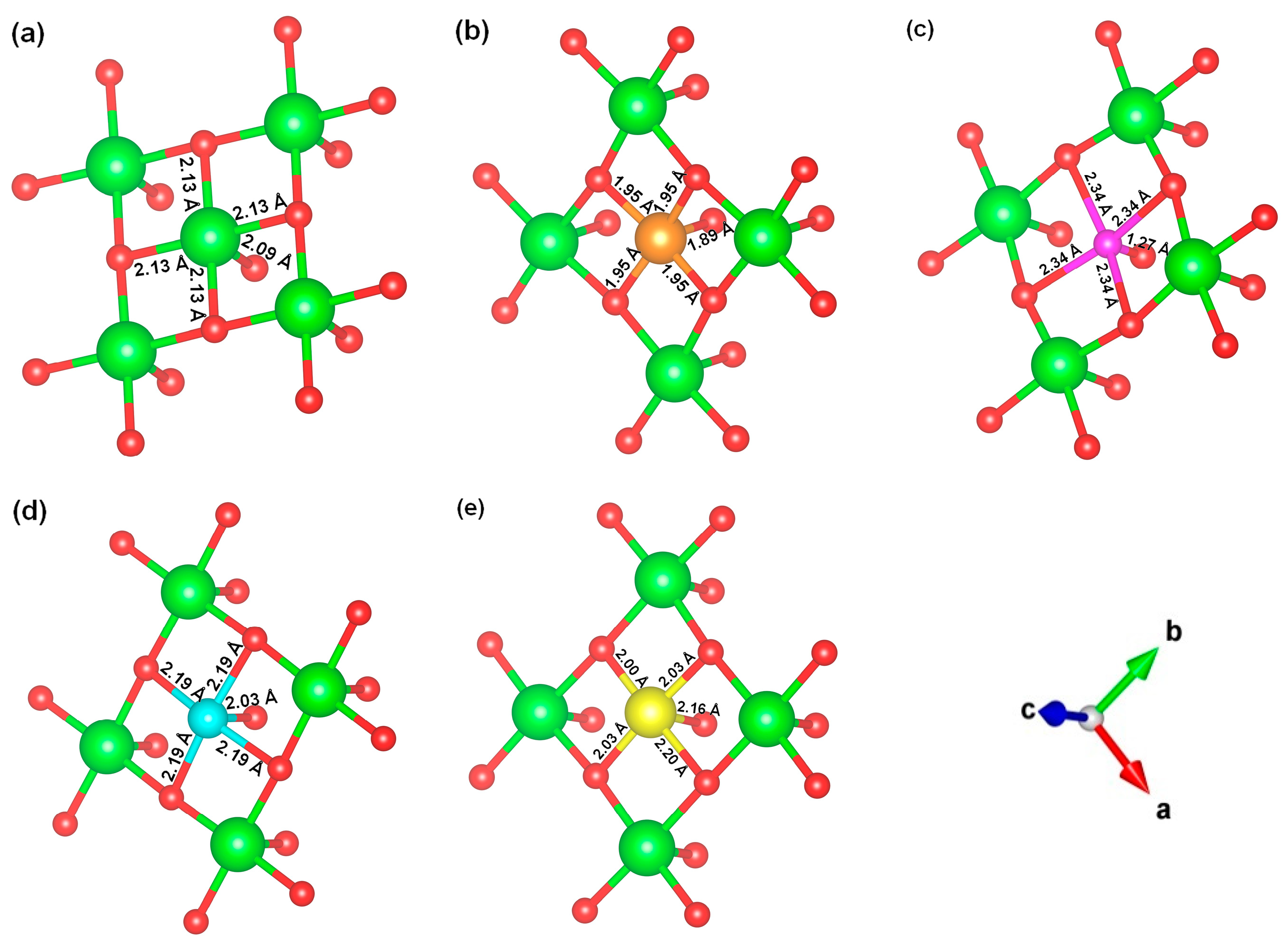
3.1. Structure of Pristine and Doped MgO Surface
3.2. Simulated CO2 Adsorption on Pristine and Doped MgO
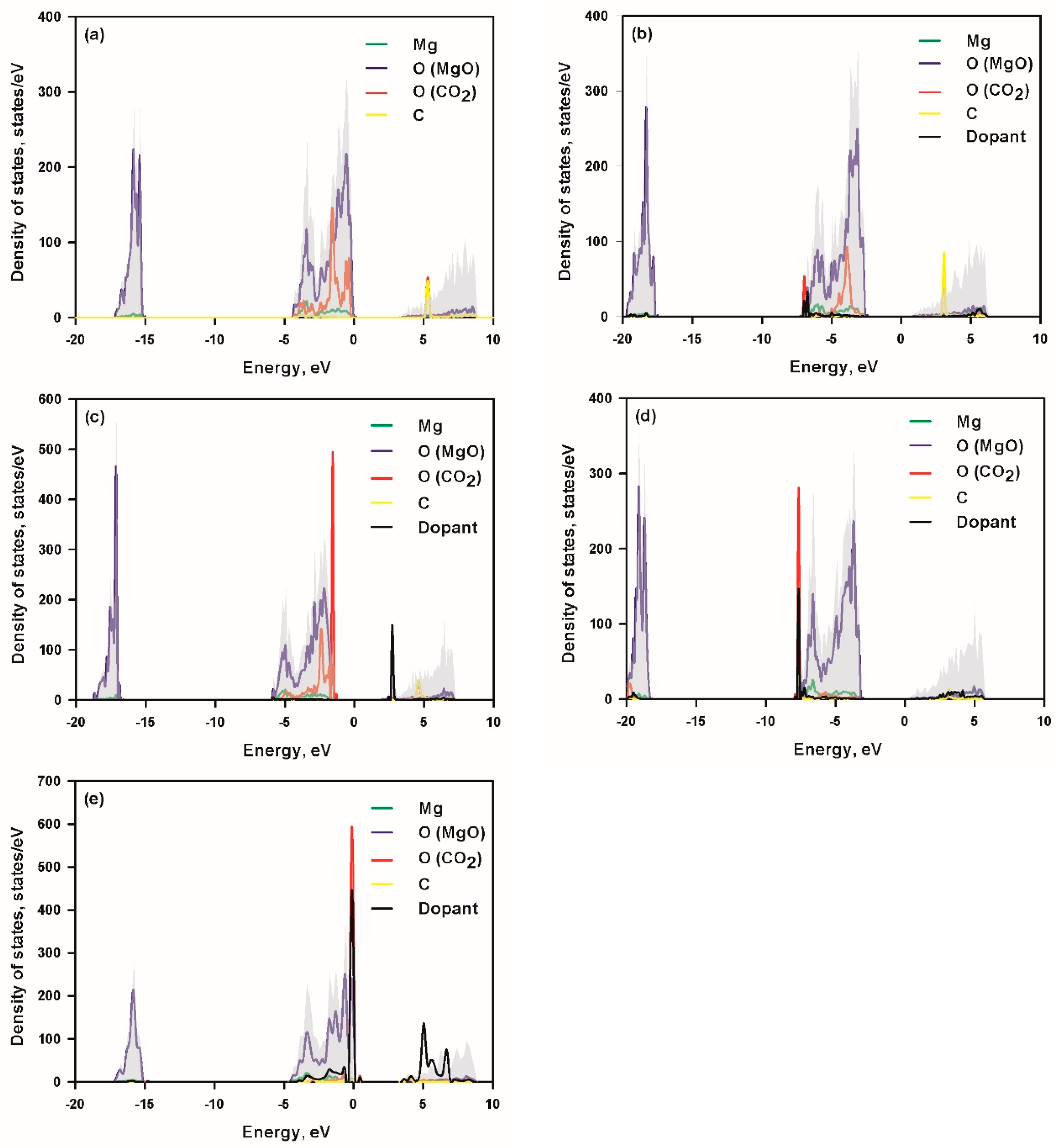
3.3. Density of States and Charge Transfer Analysis
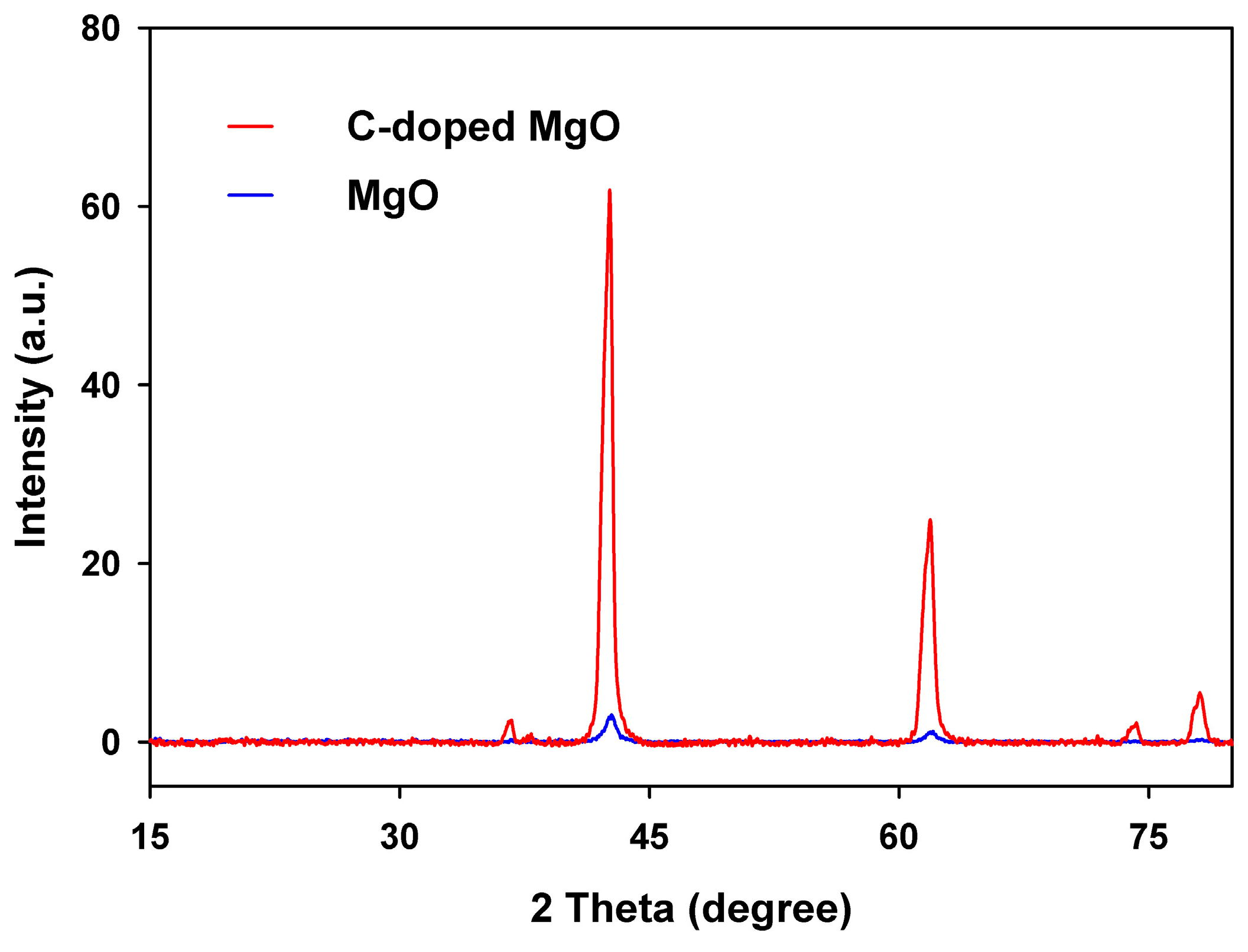
3.4. Characterization of Pristine and C-Doped MgO
3.5. Measured CO2 Adsorption on Pristine and C-Doped MgO
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IPCC. Climate Change 2021: The Physical Science Basis. In Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S.L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M.I., et al., Eds.; Cambridge University Press: Cambridge, UK, 2021; p. 2391. [Google Scholar]
- Zeng, H.; Qu, X.; Xu, D.; Luo, Y. Porous Adsorption Materials for Carbon Dioxide Capture in Industrial Flue Gas. Front. Chem. 2022, 10, 939701. [Google Scholar] [CrossRef]
- Zhu, Z.; Shi, X.; Rao, Y.; Huang, Y. Recent progress of MgO-based materials in CO2 adsorption and conversion: Modification methods, reaction condition, and CO2 hydrogenation. Chin. Chem. Lett. 2024, 35, 108954. [Google Scholar] [CrossRef]
- Li, Y.Y.; Wan, M.M.; Sun, X.D.; Zhou, J.; Wang, Y.; Zhu, J.H. Novel fabrication of an efficient solid base: Carbon-doped MgO-ZnO composite and its CO2 capture at 473 K. J. Mater. Chem. A 2015, 3, 18535–18545. [Google Scholar] [CrossRef]
- Song, G.; Ding, Y.D.; Zhu, X.; Liao, Q. Carbon dioxide adsorption characteristics of synthesized MgO with various porous structures achieved by varying calcination temperature. Colloids Surf. A-Physicochem. Eng. Asp. 2015, 470, 39–45. [Google Scholar] [CrossRef]
- Parker, A.R.; Lawrence, C.R. Water capture by a desert beetle. Nature 2001, 414, 33–34. [Google Scholar] [CrossRef]
- Senevirathna, H.L.; Wu, S.; Lee, W.P.C.; Wu, P. Morphology Design and Fabrication of Bio-Inspired Nano-MgO–Mg(OH)2 via Vapor Steaming to Enable Bulk CO2 Diffusion and Capture. Materials 2022, 15, 680. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.P.; Wen, C.; Li, Z.H. Clean production of porous MgO by thermal decomposition of Mg(OH)(2) using fluidized bed: Optimization for CO2 adsorption. J. Taiwan Inst. Chem. Eng. 2016, 63, 170–179. [Google Scholar] [CrossRef]
- Guo, X.; Ding, J.; Wu, Y.; Zhang, J.; Guo, G. Feasible fabrication of highly dispersed La2O3 promoted MgO composites for CO2 capture at mid-temperature. Mater. Chem. Phys. 2022, 279, 125734. [Google Scholar] [CrossRef]
- Liu, M.Y.; Vogt, C.; Chaffee, A.L.; Chang, S.L.Y. Nanoscale Structural Investigation of Cs2CO3-Doped MgO Sorbent for CO2 Capture at Moderate Temperature. J. Phys. Chem. C 2013, 117, 17514–17520. [Google Scholar] [CrossRef]
- Elvira, G.B.; Francisco, G.C.; Victor, S.M.; Alberto, M.L.R. MgO-based adsorbents for CO2 adsorption: Influence of structural and textural properties on the CO2 adsorption performance. J. Environ. Sci. 2017, 57, 418–428. [Google Scholar] [CrossRef]
- Niu, X.D.; Feng, Y.Y.; Xu, Y.H.; Yang, W. Synthesis of hollow Al-doped MgO spheres via a sacrificial templating method for enhanced CO2 adsorption. J. Nat. Gas Sci. Eng. 2021, 88, 10. [Google Scholar] [CrossRef]
- Hiremath, V.; Trivino, M.L.T.; Shavi, R.; Gebresillase, M.N.; Seo, J.G. Sacrificial templating method for fabrication of MgO-Al2O3@C spheres and their application to CO2 capture. Mater. Lett. 2018, 211, 304–307. [Google Scholar] [CrossRef]
- Li, Y.Y.; Dong, X.Y.M.; Sun, X.D.; Wang, Y.; Zhu, J.H. New Solid-Base Cu-MgO for CO2 Capture at 473 K and Removal of Nitrosamine. Acs Appl. Mater. Interfaces 2016, 8, 30193–30204. [Google Scholar] [CrossRef]
- Jiao, X.; Li, L.; Li, H.G.; Xiao, F.K.; Zhao, N.; Wei, W.; Zhang, B.S. Influence of preparation parameters on the structural properties and the CO2 capture performance of Mg-Zr solid sorbent. Mater. Res. Bull. 2015, 64, 163–170. [Google Scholar] [CrossRef]
- Kwak, J.S.; Kim, K.Y.; Oh, K.R.; Kwon, Y.U. Performance enhancement of all-solid CO2 absorbent based on Na2CO3-promoted MgO by using ZrO2 dispersant. Int. J. Greenh. Gas Control 2019, 81, 38–43. [Google Scholar] [CrossRef]
- Tosoni, S.; Spinnato, D.; Pacchioni, G. DFT Study of CO2 Activation on Doped and Ultrathin MgO Films. J. Phys. Chem. C 2015, 119, 27594–27602. [Google Scholar] [CrossRef]
- Hu, P.; Wang, S.; Zhuo, Y. CO2 adsorption enhancement over alkaline metal-promoted MgO with SO2, O2, and H2O present: A theoretical study. Sep. Purif. Technol. 2022, 284, 120253. [Google Scholar] [CrossRef]
- Jang, J.M.; Kang, S.G. Understanding CO2 Adsorption on a M-1 (M-2)-Promoted (Doped) MgO-CaO(100) Surface (M-1 = Li, Na, K, and Rb, M-2 = Sr): A DFT Theoretical Study. Acs Sustain. Chem. Eng. 2019, 7, 16979–16984. [Google Scholar] [CrossRef]
- Mazheika, A.; Levchenko, S.V. Ni Substitutional Defects in Bulk and at the (001) Surface of MgO from First-Principles Calculations. J. Phys. Chem. C 2016, 120, 26934–26944. [Google Scholar] [CrossRef]
- Hu, P.; Wang, S.; Zhuo, Y. Research on CO2 adsorption performances of metal-doped (Ca, Fe and Al) MgO. Sep. Purif. Technol. 2021, 276, 119323. [Google Scholar] [CrossRef]
- Hu, P.; Wang, S.; Zhuo, Y. Strengthened CO2 adsorption over Ce/Al-promoted MgO for fast capture. Sep. Purif. Technol. 2022, 287, 120518. [Google Scholar] [CrossRef]
- Manz, T.A.; Limas, N.G. Introducing DDEC6 atomic population analysis: Part 1. Charge partitioning theory and methodology. Rsc Adv. 2016, 6, 47771–47801. [Google Scholar] [CrossRef]
- Limas, N.G.; Manz, T.A. Introducing DDEC6 atomic population analysis: Part 2. Computed results for a wide range of periodic and nonperiodic materials. Rsc Adv. 2016, 6, 45727–45747. [Google Scholar] [CrossRef]
- Manz, T.A. Introducing DDEC6 atomic population analysis: Part 3. Comprehensive method to compute bond orders. Rsc Adv. 2017, 7, 45552–45581. [Google Scholar] [CrossRef]
- Limas, N.G.; Manz, T.A. Introducing DDEC6 atomic population analysis: Part 4. Efficient parallel computation of net atomic charges, atomic spin moments, bond orders, and more. Rsc Adv. 2018, 8, 2678–2707. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Norskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 19. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Wu, S.; Tan, B.T.; Senevirathna, H.L.; Wu, P. Polarization of CO2 for improved CO2 adsorption by MgO and Mg(OH)2. Appl. Surf. Sci. 2021, 562, 150187. [Google Scholar] [CrossRef]
- Hayun, S.; Tran, T.; Ushakov, S.V.; Thron, A.M.; van Benthem, K.; Navrotsky, A.; Castro, R.H.R. Experimental Methodologies for Assessing the Surface Energy of Highly Hygroscopic Materials: The Case of Nanocrystalline Magnesia. J. Phys. Chem. C 2011, 115, 23929–23935. [Google Scholar] [CrossRef]
- Chargemol. Available online: http://sourceforge.net/projects/ddec/ (accessed on 21 July 2021).
- Rumble, J. (Ed.) CRC Handbook of Chemistry and Physics, 102 ed.; CRC Press: Boca Raton, FL, USA, 2021. [Google Scholar]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Niedermaier, M.; Schwab, T.; Dolcet, P.; Bernardi, J.; Gross, S.; Bockstedte, M.; Diwald, O. Cobalt and Iron Ions in MgO Nanocrystals: Should They Stay or Should They Go. J. Phys. Chem. C 2019, 123, 25991–26004. [Google Scholar] [CrossRef]
- Hao, X.; Wang, B.; Wang, Q.; Zhang, R.; Li, D. Insight into both coverage and surface structure dependent CO adsorption and activation on different Ni surfaces from DFT and atomistic thermodynamics. Phys. Chem. Chem. Phys. 2016, 18, 17606–17618. [Google Scholar] [CrossRef]
- Ranjan, P.; Saptal, V.B.; Bera, J.K. Recent Advances in Carbon Dioxide Adsorption, Activation and Hydrogenation to Methanol using Transition Metal Carbides. ChemSusChem 2022, 15, e202201183. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Han, J.W.; Lee, K.S.; Lee, W.B. Promoting alkali and alkaline-earth metals on MgO for enhancing CO2 capture by first-principles calculations. Phys. Chem. Chem. Phys. 2014, 16, 24818–24823. [Google Scholar] [CrossRef]
- Lv, G.; Zhu, C.; Zhang, H.; Su, Y.; Qian, P. Mechanism of CO2 adsorption on point-defective MgO surfaces: First-principles study. Appl. Surf. Sci. 2022, 604, 154647. [Google Scholar] [CrossRef]
- Pan, Y.-X.; Liu, C.-J.; Wiltowski, T.S.; Ge, Q. CO2 adsorption and activation over γ-Al2O3-supported transition metal dimers: A density functional study. Catal. Today 2009, 147, 68–76. [Google Scholar] [CrossRef]
- Do, H.; Truong, H.B. Ni, Co, Zn, and Cu metal-organic framework-based nanomaterials for electrochemical reduction of CO2: A review. Beilstein J. Nanotechnol. 2023, 14, 904–911. [Google Scholar] [CrossRef]
- Wang, L.; Hou, T.; Li, Y.; Lu, H.; Gao, L. Lubrication Performances of Carbon-Doped MoSe2 Nanoparticles and Their Biocompatibility Characterization In Vitro. Front. Chem. 2021, 8, 580151. [Google Scholar] [CrossRef] [PubMed]
- Jerin, I.; Rahman, M.A.; Khan, A.H.; Hossain, M.M. Photocatalytic degradation of methylene blue under visible light using carbon-doped titanium dioxide as photocatalyst. Desalination Water Treat. 2024, 320, 100711. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dopant | Ef, eV | Charge Transfer Capacity |
|---|---|---|
| - * | 0.54 | |
| Al | 1.24 | 1.30 |
| C | 9.07 | 3.70 |
| Si | 3.64 | 3.37 |
| Ti | 2.04 | 2.54 |
| Dopant | Initial Configuration | Most Stable Structure |
|---|---|---|
| - |  | 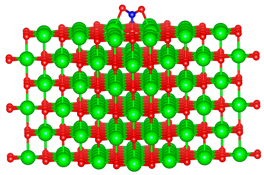 |
| Al | 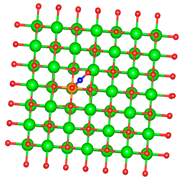 | 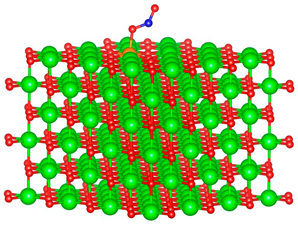 |
| C | 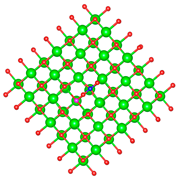 | 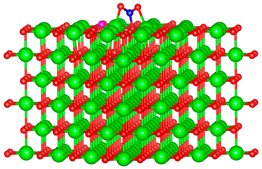 |
| Si | 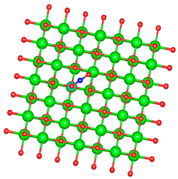 | 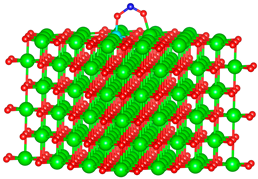 |
| Ti | 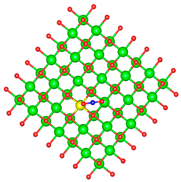 | 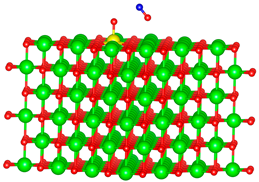 |
| Dopant | Ead | Distance, Å | CO2 Molecule | ||
|---|---|---|---|---|---|
| <OCO, deg | dC-O *, Å | dC-O *, Å | |||
| - | −0.72 | 1.44 | 132.61 | 1.26 | 1.26 |
| Al | −0.99 | 1.83 | 134.00 | 1.24 | 1.26 |
| C | −2.77 | 1.42 | 129.35 | 1.26 | 1.27 |
| Si | −1.39 | 1.63 | 116.37 | 1.27 | 1.42 |
| Ti | −2.80 | n/a | n/a | n/a | n/a |
| Dopant | Charge Transfer Capacity | CO2 | O1 | O2 |
|---|---|---|---|---|
| - | 0.56 | −0.50 | −0.63 | −0.63 |
| Al | 1.29 | −0.71 | −0.52 | −0.52 |
| C | 3.79 | −0.53 | −0.65 | −0.68 |
| Si | 2.37 | −1.18 | −0.59 | −0.60 |
| Ti | 2.00 | −0.91 | −0.20 | −0.88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.; Lee, W.P.C.; Thenuwara, H.N.; Li, X.; Wu, P. Enhancing CO2 Adsorption on MgO: Insights into Dopant Selection and Mechanistic Pathways. Biomimetics 2025, 10, 9. https://doi.org/10.3390/biomimetics10010009
Wu S, Lee WPC, Thenuwara HN, Li X, Wu P. Enhancing CO2 Adsorption on MgO: Insights into Dopant Selection and Mechanistic Pathways. Biomimetics. 2025; 10(1):9. https://doi.org/10.3390/biomimetics10010009
Chicago/Turabian StyleWu, Shunnian, W. P. Cathie Lee, Hashan N. Thenuwara, Xu Li, and Ping Wu. 2025. "Enhancing CO2 Adsorption on MgO: Insights into Dopant Selection and Mechanistic Pathways" Biomimetics 10, no. 1: 9. https://doi.org/10.3390/biomimetics10010009
APA StyleWu, S., Lee, W. P. C., Thenuwara, H. N., Li, X., & Wu, P. (2025). Enhancing CO2 Adsorption on MgO: Insights into Dopant Selection and Mechanistic Pathways. Biomimetics, 10(1), 9. https://doi.org/10.3390/biomimetics10010009