
Future of Dutch NGS-Based Newborn Screening: Exploring the Technical Possibilities and Assessment of a Variant Classification Strategy

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Setup
2.2. Sample Collection and DNA Extraction
2.3. Preparation of Samples, Sequencing, Data Filtering, and Classification
2.4. Outcome of the Study
- Technical performance of tNGS, WES, and WGS. The number of failed samples, overall coverage, and specific regions not covered >20x were reported.
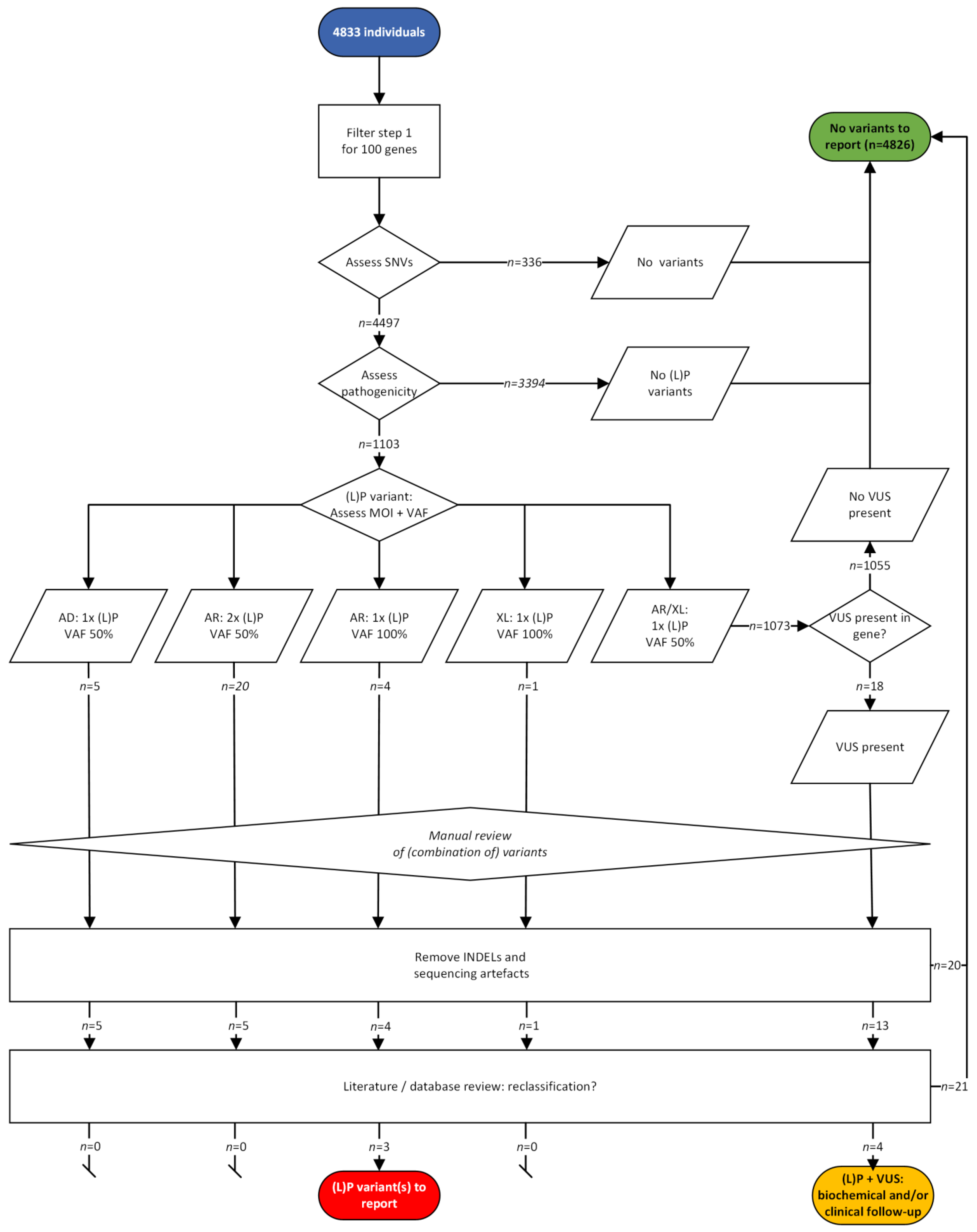
- Outcomes of the two variant filtering strategies. First, we measured the number of variants using the strict filtering strategy reporting only P and LP variants ((L)P) filter strategy; Figure 2B, left). We then calculated the number of true positives (TP), true negatives (TN), FP and false negatives (FN) based on the definitions described in Supplementary File S1. Second, we tested a filtering strategy with an additional step (Figure 2B, right). When only one P or LP variant was found in an AR or XLR gene, we also reported VUS found in the same gene (extra VUS filter strategy). Here, we used the same definitions for TP/FP/FN/TN, but the samples in which one LP or P variant and a VUS were detected, were now considered positive. Carriership was defined as presence of one LP or P variant detected with a VAF of 50% in an AR gene in any sample.
- Estimated turnaround time. For a series of 96 samples, we measured the time needed to obtain results. We included the time needed for automated sample preparation, sequencing, data processing, and data analysis. For tNGS data analysis, an automated variant interpretation pipeline was used to obtain relevant variants for each sample (https://github.com/molgenis/vip, accessed on 1 May 2022). For WES and WGS, downstream processing was performed using an automated data analysis pipeline and custom-made annotation [20], with a bioinformatic filter for the selected genes.
3. Results
3.1. Technical Performance
3.2. Filter Strategy Assessment
3.3. Carriership
3.4. Turnaround Time
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.C.; Adams, J. Current Status of Newborn Screening Worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Castiñeras, D.E.; Couce, M.-L.; Marín, J.L.; González-Lamuño, D.; Rocha, H. Newborn Screening for Metabolic Disorders in Spain and Worldwide. An. Pediatría 2019, 91, 128e.1–128e.14. [Google Scholar] [CrossRef]
- van Dijk, T.; Kater, A.; Jansen, M.; Dondorp, W.J.; Blom, M.; Kemp, S.; Langeveld, M.; Cornel, M.C.; van der Pal, S.M.; Henneman, L. Expanding Neonatal Bloodspot Screening: A Multi-Stakeholder Perspective. Front. Pediatr. 2021, 9, 706394. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Loarte, T.; Thompson, J.D.; Merritt, J.L. Considering Proximal Urea Cycle Disorders in Expanded Newborn Screening. Int. J. Neonatal Screen. 2020, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Stroek, K.; Boelen, A.; Bouva, M.J.; De Sain-van der Velden, M.; Schielen, P.C.J.I.; Maase, R.; Engel, H.; Jakobs, B.; Kluijtmans, L.A.J.; Mulder, M.F.; et al. Evaluation of 11 Years of Newborn Screening for Maple Syrup Urine Disease in the Netherlands and a Systematic Review of the Literature: Strategies for Optimization. JIMD Rep. 2020, 54, 68–78. [Google Scholar] [CrossRef]
- Derks, T.G.J.; Boer, T.S.; Assen, A.; Bos, T.; Ruiter, J.; Waterham, H.R.; Niezen-Koning, K.E.; Wanders, R.J.A.; Rondeel, J.M.M.; Loeber, J.G.; et al. Neonatal Screening for Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency in The Netherlands: The Importance of Enzyme Analysis to Ascertain True MCAD Deficiency. J. Inherit. Metab. Dis. 2008, 31, 88–96. [Google Scholar] [CrossRef]
- French, C.E.; Delon, I.; Dolling, H.; Sanchis-Juan, A.; Shamardina, O.; Mégy, K.; Abbs, S.; Austin, T.; Bowdin, S.; Branco, R.G.; et al. Whole Genome Sequencing Reveals That Genetic Conditions Are Frequent in Intensively Ill Children. Intensive Care Med. 2019, 45, 627–636. [Google Scholar] [CrossRef]
- Meng, L.; Pammi, M.; Saronwala, A.; Magoulas, P.; Ghazi, A.R.; Vetrini, F.; Zhang, J.; He, W.; Dharmadhikari, A.V.; Qu, C.; et al. Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA Pediatr. 2017, 171, e173438. [Google Scholar] [CrossRef]
- de Ligt, J.; Willemsen, M.H.; van Bon, B.W.M.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef]
- Neveling, K.; Feenstra, I.; Gilissen, C.; Hoefsloot, L.H.; Kamsteeg, E.J.; Mensenkamp, A.R.; Rodenburg, R.J.T.; Yntema, H.G.; Spruijt, L.; Vermeer, S.; et al. A Post-Hoc Comparison of the Utility of Sanger Sequencing and Exome Sequencing for the Diagnosis of Heterogeneous Diseases. Hum. Mutat. 2013, 34, 1721–1726. [Google Scholar] [CrossRef]
- Olde Keizer, R.A.C.M.; Marouane, A.; Kerstjens-Frederikse, W.S.; Deden, A.C.; Lichtenbelt, K.D.; Jonckers, T.; Vervoorn, M.; Vreeburg, M.; Henneman, L.; de Vries, L.S.; et al. Rapid Exome Sequencing as a First-Tier Test in Neonates with Suspected Genetic Disorder: Results of a Prospective Multicenter Clinical Utility Study in the Netherlands. Eur. J. Pediatr. 2023, 182, 2683–2692. [Google Scholar] [CrossRef]
- Holm, I.A.; Agrawal, P.B.; Ceyhan-Birsoy, O.; Christensen, K.D.; Fayer, S.; Frankel, L.A.; Genetti, C.A.; Krier, J.B.; LaMay, R.C.; Levy, H.L.; et al. The BabySeq Project: Implementing Genomic Sequencing in Newborns. BMC Pediatr. 2018, 18, 225. [Google Scholar] [CrossRef]
- van Campen, J.C.; Sollars, E.S.A.; Thomas, R.C.; Bartlett, C.M.; Milano, A.; Parker, M.D.; Dawe, J.; Winship, P.R.; Peck, G.; Grafham, D.; et al. Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service. Int. J. Neonatal Screen. 2019, 5, 40. [Google Scholar] [CrossRef]
- Burton, B.K.; Veldman, A.; Kiewiet, M.B.G.; Rebecca Heiner-Fokkema, M.; Nelen, M.R.; Sinke, R.J.; Sikkema-Raddatz, B.; Voorhoeve, E.; Westra, D.; Dollé, M.E.T.; et al. Neonatal Screening Towards Next-Generation Sequencing (NGS)-Based Newborn Screening: A Technical Study to Prepare for the Challenges Ahead. Int. J. Neonatal Screen. 2022, 8, 17. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Bick, D.; Scott, R.; Hopkins, H.; Krones, T.; Gross, E.S.; Bonham, J.R. Genomic Newborn Screening: Are We Entering a New Era of Screening? J. Inherit. Metab. Dis. 2023, 46, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Gregg, A.R.; Aarabi, M.; Klugman, S.; Leach, N.T.; Bashford, M.T.; Goldwaser, T.; Chen, E.; Sparks, T.N.; Reddi, H.V.; Rajkovic, A.; et al. Screening for Autosomal Recessive and X-Linked Conditions during Pregnancy and Preconception: A Practice Resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, S.F.; Smith, L.D.; Kunard, C.M.; Bainbridge, M.; Batalov, S.; Benson, W.; Blincow, E.; Caylor, S.; Chambers, C.; del Angel, G.; et al. A Genome Sequencing System for Universal Newborn Screening, Diagnosis, and Precision Medicine for Severe Genetic Diseases. Am. J. Hum. Genet. 2022, 109, 1605–1619. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.S.; Wilkinson, M.F.; Gecz, J. Nonsense-Mediated MRNA Decay: Inter-Individual Variability and Human Disease. Neurosci. Biobehav. Rev. 2014, 46, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.J.; De Vries, P.; De Vries, B.B.A.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-Analysis of 2104 Trios Provides Support for 10 New Genes for Intellectual Disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef]
- Morales, J.; Pujar, S.; Loveland, J.E.; Astashyn, A.; Bennett, R.; Berry, A.; Cox, E.; Davidson, C.; Ermolaeva, O.; Farrell, C.M.; et al. A Joint NCBI and EMBL-EBI Transcript Set for Clinical Genomics and Research. Nature 2022, 604, 310–315. [Google Scholar] [CrossRef]
- Hwang, J.; Jaroensuk, J.; Leimanis, M.L.; Russell, B.; McGready, R.; Day, N.; Snounou, G.; Nosten, F.; Imwong, M. Long-Term Storage Limits PCR-Based Analyses of Malaria Parasites in Archival Dried Blood Spots. Malar. J. 2012, 11, 339. [Google Scholar] [CrossRef]
- Tangeraas, T.; Sæves, I.; Klingenberg, C.; Jørgensen, J.; Kristensen, E.; Gunnarsdottir, G.; Hansen, E.V.; Strand, J.; Lundman, E.; Ferdinandusse, S.; et al. Performance of Expanded Newborn Screening in Norway Supported by Post-Analytical Bioinformatics Tools and Rapid Second-Tier DNA Analyses. Int. J. Neonatal Screen. 2020, 6, 51. [Google Scholar] [CrossRef]
- Malvagia, S.; Forni, G.; Ombrone, D.; la Marca, G. Development of Strategies to Decrease False Positive Results in Newborn Screening. Int. J. Neonatal Screen. 2020, 6, 84. [Google Scholar] [CrossRef]
- Monitor van de Neonatale Hielprikscreening 2021 | Prenatale En Neonatale Screeningen. Available online: https://www.pns.nl/documenten/monitor-van-neonatale-hielprikscreening-2021 (accessed on 21 May 2023).
- Karaceper, M.D.; Chakraborty, P.; Coyle, D.; Wilson, K.; Kronick, J.B.; Hawken, S.; Davies, C.; Brownell, M.; Dodds, L.; Feigenbaum, A.; et al. The Health System Impact of False Positive Newborn Screening Results for Medium-Chain Acyl-CoA Dehydrogenase Deficiency: A Cohort Study. Orphanet J. Rare Dis. 2016, 11, 12. [Google Scholar] [CrossRef]
- Lund, A.M.; Wibrand, F.; Skogstrand, K.; Bækvad-Hansen, M.; Gregersen, N.; Andresen, B.S.; Hougaard, D.M.; Dunø, M.; Olsen, R.K.J. Use of Molecular Genetic Analyses in Danish Routine Newborn Screening. Int. J. Neonatal Screen. 2021, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.H.; Zhang, T.; Ceyhan-Birsoy, O.; Genetti, C.A.; Lebo, M.S.; Yu, T.W.; Parad, R.B.; Holm, I.A.; Rehm, H.L.; Beggs, A.H.; et al. Discordant Results between Conventional Newborn Screening and Genomic Sequencing in the BabySeq Project. Genet. Med. 2021, 23, 1372–1375. [Google Scholar] [CrossRef]
- la Marca, G.; Carling, R.S.; Moat, S.J.; Yahyaoui, R.; Ranieri, E.; Bonham, J.R.; Schielen, P.C.J.I. Current State and Innovations in Newborn Screening: Continuing to Do Good and Avoid Harm. Int. J. Neonatal Screen. 2023, 9, 15. [Google Scholar] [CrossRef]
- Narravula, A.; Garber, K.B.; Askree, S.H.; Hegde, M.; Hall, P.L. Variants of Uncertain Significance in Newborn Screening Disorders: Implications for Large-Scale Genomic Sequencing. Genet. Med. 2017, 19, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Bick, D.; Ahmed, A.; Deen, D.; Ferlini, A.; Garnier, N.; Kasperaviciute, D.; Leblond, M.; Pichini, A.; Rendon, A.; Satija, A.; et al. Newborn Screening by Genomic Sequencing: Opportunities and Challenges. Int. J. Neonatal Screen. 2022, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, B.; Christensen, K.D.; Genetti, C.A.; Parad, R.B.; Robinson, J.O.; Blout Zawatsky, C.L.; Zettler, B.; Beggs, A.H.; Holm, I.A.; Green, R.C.; et al. Parental Attitudes toward Standard Newborn Screening and Newborn Genomic Sequencing: Findings from the BabySeq Study. Front. Genet. 2022, 13, 867371. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, A.J.; Lloyd-Puryear, M.; Brosco, J.P.; Therrell, B.; Bush, L.; Berry, S.; Brower, A.; Bonhomme, N.; Bowdish, B.; Chrysler, D.; et al. Including ELSI Research Questions in Newborn Screening Pilot Studies. Genet. Med. 2019, 21, 525–533. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Variant | Detected? | ||
|---|---|---|---|---|
| tNGS | WES | WGS | ||
| 1 | SLC2A2 Chr3(GRCh37):g.170716187T>C NM_000340.3:c.1771-2A>G p.?; homozygous | Yes | Yes | Yes |
| 2 | GCDH Chr19(GRCh37):g.13002736del NM_000159.4:c.219del p.(Tyr74fs); heterozygous | Yes | No * | No * |
| GCDH Chr19(GRCh37):g.13004444G>A NM_000159.4:c.482G>A p.(Arg161Gln); heterozygous | Yes | Yes | Yes | |
| 3 | CBS Chr21(GRCh37):g.44478972C>T NM_000071.3:c.1330G>A p.(Asp444Asn); heterozygous | Yes | Yes | No |
| CBS Chr21(GRCh37):g.44484032_4484034del NM_000071.3:c.805_807del p.(Lys269del); heterozygous | Yes | Yes | No | |
| 4 | PCCB Chr3(GRCh37):g.136035806dup NM_001178014.2:c.1050dup p.(Glu351*); heterozygous | Yes | No data | Yes |
| PCCB Chr3(GRCh37):g.136046016_136046029delinsTAGAGCACAGGA NM_001178014.2:c.1278_1291delinsTAGAGCACAGGA p.(Gly427fs); heterozygous | Yes | No data | Yes | |
| 5 | PAH Chr12(GRCh37):g.103234177C>T NM_000277.3:c.1315+1G>A p.?; het | Yes | Yes | Yes |
| PAH Chr12(GRCh37):g.103288604G>T NM_000277.3:c.261C>A p.(Ser87Arg); heterozygous | Yes | Yes | Yes | |
| 6 | SLC22A5 Chr5(GRCh37):g.131705516G>A NM_003060.4:c.-149G>A p.?; heterozygous | Yes | No | No |
| SLC22A5 Chr5(GRCh37):g.131719951G>A NM_003060.4:c.610G>A p.(Gly204Ser); heterozygous | No * | No | No | |
| 7 | SLC2A1 Chr1(GRCh37):g.43395707C>G NM_006516.4:c.517-1G>C p.?; heterozygous | Yes | Yes | Yes |
| 8 | PCCA Chr13(GRCh37):g.100888120G>C NM_000282.4:c.625G>C p.(Ala209Pro); heterozygous | No * | No * | No * |
| PCCA Chr13(GRCh37):g.100925458dup NM_000282.4:c.923dup p.(Leu308fs); heterozygous | Yes | Yes | Yes | |
| 9 | OAT Chr10(GRCh37):g.126089510C>T NM_000274.4:c.1058G>A p.(Gly353Asp); heterozygous | Yes | Yes | Yes |
| OAT Chr10(GRCh37):g.126086661del NM_000274.4:c.1171del p.(Trp391fs); heterozygous | Yes | Yes | Yes | |
| 10 | ACADM Chr1(GRCh37):g.76198409T>C NM_000016.6:c.199T>C p.(Tyr67His); heterozygous | Yes | Yes | Yes |
| ACADM Chr1(GRCh37):g.76226846A>G NM_000016.6:c.985A>G p.(Lys329Glu); heterozygous | Yes | Yes | Yes | |
| 11 | SLC52A3 Chr20(GRCh37):g.744576G>C NM_033409.4:c.639C>G p.(Tyr213*); heterozygous | Yes | Yes | Yes |
| SLC52A3 Chr20(GRCh37):g.744542_744544del NM_033409.4:c.678_680del p.(Leu227del); heterozygous | No * | No * | No * | |
| 12 | FAH Chr15(GRCh37):g.80464558T>G NM_000137.4:c.674T>G p.(Ile225Ser); heterozygous | No * | No * | No * |
| FAH Chr15(GRCh37):g.80472572G>A NM_000137.4:c.1062+5G>A p.?; heterozygous | Yes | Yes | Yes | |
| 13 | ASS1 Chr9(GRCh37):g.133352345_133352352del NM_054012.4:c.685_688+4del p.(fs232*); heterozygous | Yes | Yes | Yes |
| ASS1 Chr9(GRCh37):g.133355813G>A NM_054012.4:c.815G>A p.(Arg272His); heterozygous | Yes | Yes | Yes | |
| 14 | BCKDHB Chr6(GRCh37):g.80982870C>T NM_183050.4:c.970C>T p.(Arg324*); homozygous | Yes | Yes | Yes |
| 15 | GAMT Chr19(GRCh37):g.1398988A>G NM_000156.6:c.497T>C p.(Leu166Pro); homozygous | Yes | Yes | Yes |
| 16 | ETFB Chr19(GRCh37):g.51848627_51848629del NM_001985.3:c.614_616delAGA p.(Lys205del); homozygous | Yes | Yes | Yes |
| 17 | MMACHC Chr1(GRCh37):g.45973222G>T NM_015506.3:c.276G>T p.(Glu92Asp); homozygous | Yes | Yes | Yes |
| 18 | ACAT1 Chr11(GRCh37):g.108010834C>T NM_000019.4:c.662C>T p.(Arg208*); heterozygous | Yes | Yes | Yes |
| ACAT1 Chr1(GRCh37):g.108016927A>C NM_000019.4:c.1006-2A>C p.?; heterozygous | Yes | Yes | Yes | |
| 19 | MMUT Chr6(GRCh37):g.49425703G>A NM_000255.4:c.454C>T p.(Arg152*); heterozygous | Yes | Yes | Yes |
| MMUT Chr6(GRCh37):g.49425502T>A NM_00255.4:c.665A>T p.(Asn219Tyr); heterozygous | Yes | Yes | Yes | |
| 20 | OTC ChrX(GRCh37):g.38271205C>T NM_000531.6:c.958C>T p.(Arg320*); homozygous | Yes | Yes | No data |
| 21 | ACADVL Chr17(GRCh37):g.7123482del NM_000018.4:c.104del p.(Pro35fs); heterozygous | Yes | Yes | Yes |
| ACADVL Chr17(GRCh37):g.7125591T>C NM_000018.4:c.848T>C p.(Val283Ala); heterozygous | Yes | Yes | Yes | |
| 22 | SLC52A2 Chr8(GRCh37):g.145583300dup NM_001363118.2:c.148dup p.(Tyr50fs); heterozygous | Yes | Yes | Yes |
| SLC52A2 Chr8(GRCh37):g.145584264T>C NM_001363118.2:c.1016T>C p.(Leu339Pro); heterozygous | Yes | Yes | Yes | |
| 23 | MMACHC Chr1(GRCh37):g.45973217dup NM_015506.3:c.271dup p.(Arg91fs); heterozygous | No data | No data | Yes |
| MMACHC Chr1(GRCh37):g.45973222G>T NM_015506.3:c.276G>T p.(Glu92Asp); heterozygous | No data | No data | Yes | |
| 24 | DNAJC12 Chr10(GRCh37):g.69583144del NM_021800.3:c.85del p.(Gln29fs); heterozygous | Yes | Yes | Yes |
| DNAJC12 Chr10(GRCh37):g.69556875C>A NM_021800.3:c.596G>T p.(*199Leuext*42); heterozygous | Yes | Yes | Yes | |
| 25 | ALDH7A1 Chr5(GRCh37):g.1288206del NM_001182.5:c.1513del p.(Ala505fs); homozygous | Yes | Yes | Yes |
| 26 | IVD Chr15(GRCh37):g.40699855A>T NM_002225.5:c.163A>T p.(Lys55*); heterozygous | Yes | Yes | Yes |
| IVD Chr15(GRCh37):g.40710350A>G NM_002225.3:c.1169A>G p.(Asp390Gly); heterozygous | No * | No * | No * | |
| 27 | HMGCL Chr1(GRCh37):g.24147022C>T NM_000191.3:c.122G>A p.(Arg41Gln); homozygous | Yes | Yes | Yes |
| 28 | Chr17(GRCh37):g.3493545_3564028del; heterozygous (57 kb deletion including CTNS gene) | Yes | Yes | Yes |
| CTNS Chr17(GRCh37):g.3543518_3543521del NM_004937.3:c.18_21del p.(Thr7fs); hemizygous | Yes | Yes | Yes | |
| 29 | G6PC Chr17(GRCh37):g.41052972dell NM_000151.4:c.79del p.(Gln27fs); heterozygous | Yes | Yes | Yes |
| G6PC Chr17(GRCh37):g.41063157del NM_000151.4:c.788del p.(Lys263fs); heterozygous | Yes | Yes | Yes | |
| 30 | ETFA Chr15(GRCh37):g.76603769C>T NM_000126.4:c.-40G>A p.?: homozygous | No | No | No |
| 31 | ABCD1 ChrX(GRCh37):g.152991164A>G NM_000033.4:c.443A>G p.(Asn148Ser); hem. | Yes | Yes | No |
| 32 | AGL Chr1(GRCh37):g.100316614C>T NM_000642.3:c.16C>T p.(Gln6*); heterozygous | No data | Yes | Yes |
| AGL Chr1(GRCh37):g.100387137dup NM_000642.3:c.4529dup p.(Tyr1510*); heterozygous | No data | Yes | Yes | |
| 33 | ASL Chr7(GRCh37):g.65551586T>C NM_000048.4:c.461T>C p.(Leu154Pro); heterozygous | Yes | No data | Yes |
| ASL Chr7(GRCh37):g.65551738G>A NM_000048.4:c.532G>A p.(Val178Met); heterozygous | Yes | No data | Yes | |
| 34 | BCKDHA Chr19(GRCh37):g.41916527T>A NM_000709.4:c.109-15T>A p.?; homozygous | No | No | No |
| 35 | BTD Chr3(GRCh37):g.15676984_15676990delinsTCC NM_000060.2:c.98_104delinsTCC p.(Cys33fs); heterozygous | Yes | Yes | Yes |
| No second variant found in diagnostic setting | n.a. | |||
| 36 | CAD Chr2(GRCh37):g.27460617C>T NM_004341.5:c.4595C>T p.(Ala1532Val); homozygous | No | No | No |
| 37 | CPT2 Chr1(GRCh37):g.53666438C>G NM_000098.3:c.200C>G p.(Ala67Gly); heterozygous | Yes | No * | No * |
| CPT2 Chr1(GRCh37):g.53676026C>T NM_000098.3:c.680C>T p.(Pro227Leu); heterozygous | Yes | Yes | Yes | |
| 38 | CYP27A1 Chr2(GRCh37):g.219677818C>T NM_000784.4:c.1016C>T p.(Thr339Met); heterozygous | Yes | Yes | Yes |
| CYP27A1 Chr2(GRCh37):g.219678909C>T NM_000784.4:c.1183C>T p.(Arg395Cys); heterozygous | Yes | Yes | Yes | |
| 39 | FOLR1 Chr11(GRCh37):g.71906952T>C NM_016729.3:c.505T>C p.(Cys169Arg); homozygous | No | Yes | Yes |
| 40 | Chr12(GRCh37):g.1955262_22837888del; heterozygous deletion including GYS2 | Yes | Yes | Yes |
| GYS2 c.495+1G>T p.?; heterozygous | Yes | Yes | Yes | |
| 41 | HADHA Chr2(GRCh37):g.26418053C>G NM_000182.5:c.1528G>C p.(Glu510Gln); heterozygous | Yes | Yes | Yes |
| HADHA Chr2(GRCh37):g.26414401del NM_000182.5:c.2099del p.(Gly700fs); heterozygous | Yes | Yes | Yes | |
| 42 | HMGCS2 Chr1(GRCh37):g.120307008G>A NM_005518.4:c.346C>T p.(Arg116Cys); heterozygous | No * | No | No data |
| HMGCS2 Chr1(GRCh37):g.120302538C>T NM_005518.4:c.634G>A p.(Gly212Arg); heterozygous | Yes | Yes | No data | |
| 43 | MCCC1 Chr3(GRCh37):g.18278896A>T NM_020166.5:c.639+2T>A p.?; homozygous | Yes | Yes | No data |
| 44 | MCCC2 Chr5(GRCh37):g.70945074C>T NM_022132.5:c.1367C>T p.(Ala456Val); heterozygous | Yes | No * | No * |
| MCCC2 Chr5(GRCh37):g.70948566A>G NM_022132.5:c.1559A>G p.(Tyr520Cys); heterozygous | Yes | Yes | Yes | |
| 45 | OXCT1 Chr5(GRCh37):g.41803250C>T NM_000436.4:c.971G>A p.(Gly324Glu); homozygous | No data | Yes | Yes |
| 46 | TH Chr11(GRCh37):g.2189135C>T NM_199292.3:c.698G>A p.(Arg233His); heterozygous | Yes | Yes | Yes |
| TH Chr11(GRCh37):g.2186980G>A NM_199292.3:c.1211C>T p.(Thr404Met); heterozygous | No * | No * | No * | |
| 47 | SLC2A1 Chr1(GRCh37):g.43395453T>A NM_006516.4:c.680-2A>T p.?; heterozygous | Yes | Yes | Yes |
| tNGS | WES | WGS | ||||||
|---|---|---|---|---|---|---|---|---|
| Sample | Variant (s) | Reason Variant Missed | Sample | Variant (s) | Reason Variant Missed | Sample | Variant (s) | Reason Variant Missed |
| 6 | SLC22A5 Chr5(GRCh37):g.131705516G>A NM_003060.4:c.-149G>A p.?; heterozygous | 3′UTR variant filtered out, but present in raw data/other variant VUS | 6 | SLC22A5 Chr5(GRCh37):g.131705516G>A NM_003060.4:c.-149G>A p.?; heterozygous | 3′UTR variant filtered out, but present in raw data/other variant VUS | |||
| 30 | ETFA Chr15(GRCh37):g.76603769C>T NM_000126.4:c.-40G>A p.?: homozygous | hom. VUS/present in raw data | 30 | ETFA Chr15(GRCh37):g.76603769C>T NM_000126.4:c.-40G>A p.?: homozygous | hom. VUS/present in raw data | 30 | ETFA Chr15(GRCh37):g.76603769C>T NM_000126.4:c.-40G>A p.?: homozygous | hom. VUS/present in raw data |
| 34 | BCKDHA Chr19(GRCh37):g.41916527T>A NM_000709.4:c.109-15T>A p.?; homozygous | hom. VUS/present in raw data | 34 | BCKDHA Chr19(GRCh37):g.41916527T>A NM_000709.4:c.109-15T>A p.?; homozygous | hom. VUS/present in raw data | 34 | BCKDHA Chr19(GRCh37):g.41916527T>A NM_000709.4:c.109-15T>A p.?; homozygous | hom. VUS/present in raw data |
| 36 | CAD Chr2(GRCh37):g.27460617C>T NM_004341.5:c.4595C>T p.(Ala1532Val); homozygous | hom. VUS/present in raw data | 36 | CAD Chr2(GRCh37):g.27460617C>T NM_004341.5:c.4595C>T p.(Ala1532Val); homozygous | hom. VUS/present in raw data | 36 | CAD Chr2(GRCh37):g.27460617C>T NM_004341.5:c.4595C>T p.(Ala1532Val); homozygous | hom. VUS/present in raw data |
| 39 | FOLR1 Chr11(GRCh37):g.71906952T>C NM_016729.3:c.505T>C p.(Cys169Arg); homozygous | hom. VUS/present in raw data | 3 | CBS Chr21(GRCh37):g.44478972C>T NM_000071.3:c.1330G>A p.(Asp444Asn); heterozygous CBS Chr21(GRCh37):g.44484032_4484034del NM_000071.3:c.805_807del p.(Lys269del); heterozygous | Low coverage and pseudogene | |||
| 31 | ABCD1 ChrX(GRCh37):g.152991164A>G NM_000033.4:c.443A>G p.(Asn148Ser); hem. | Low coverage and pseudogene | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiewiet, G.; Westra, D.; de Boer, E.N.; van Berkel, E.; Hofste, T.G.J.; van Zweeden, M.; Derks, R.C.; Leijsten, N.F.A.; Ruiterkamp-Versteeg, M.H.A.; Charbon, B.; et al. Future of Dutch NGS-Based Newborn Screening: Exploring the Technical Possibilities and Assessment of a Variant Classification Strategy. Int. J. Neonatal Screen. 2024, 10, 20. https://doi.org/10.3390/ijns10010020
Kiewiet G, Westra D, de Boer EN, van Berkel E, Hofste TGJ, van Zweeden M, Derks RC, Leijsten NFA, Ruiterkamp-Versteeg MHA, Charbon B, et al. Future of Dutch NGS-Based Newborn Screening: Exploring the Technical Possibilities and Assessment of a Variant Classification Strategy. International Journal of Neonatal Screening. 2024; 10(1):20. https://doi.org/10.3390/ijns10010020
Chicago/Turabian StyleKiewiet, Gea, Dineke Westra, Eddy N. de Boer, Emma van Berkel, Tom G. J. Hofste, Martine van Zweeden, Ronny C. Derks, Nico F. A. Leijsten, Martina H. A. Ruiterkamp-Versteeg, Bart Charbon, and et al. 2024. "Future of Dutch NGS-Based Newborn Screening: Exploring the Technical Possibilities and Assessment of a Variant Classification Strategy" International Journal of Neonatal Screening 10, no. 1: 20. https://doi.org/10.3390/ijns10010020
APA StyleKiewiet, G., Westra, D., de Boer, E. N., van Berkel, E., Hofste, T. G. J., van Zweeden, M., Derks, R. C., Leijsten, N. F. A., Ruiterkamp-Versteeg, M. H. A., Charbon, B., Johansson, L., Bos-Kruizinga, J., Veenstra, I. J., de Sain-van der Velden, M. G. M., Voorhoeve, E., Heiner-Fokkema, M. R., van Spronsen, F., Sikkema-Raddatz, B., & Nelen, M. (2024). Future of Dutch NGS-Based Newborn Screening: Exploring the Technical Possibilities and Assessment of a Variant Classification Strategy. International Journal of Neonatal Screening, 10(1), 20. https://doi.org/10.3390/ijns10010020