

Establishment of a Pilot Newborn Screening Program for Spinal Muscular Atrophy in Saint Petersburg †

 ,
,  , ,
, ,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
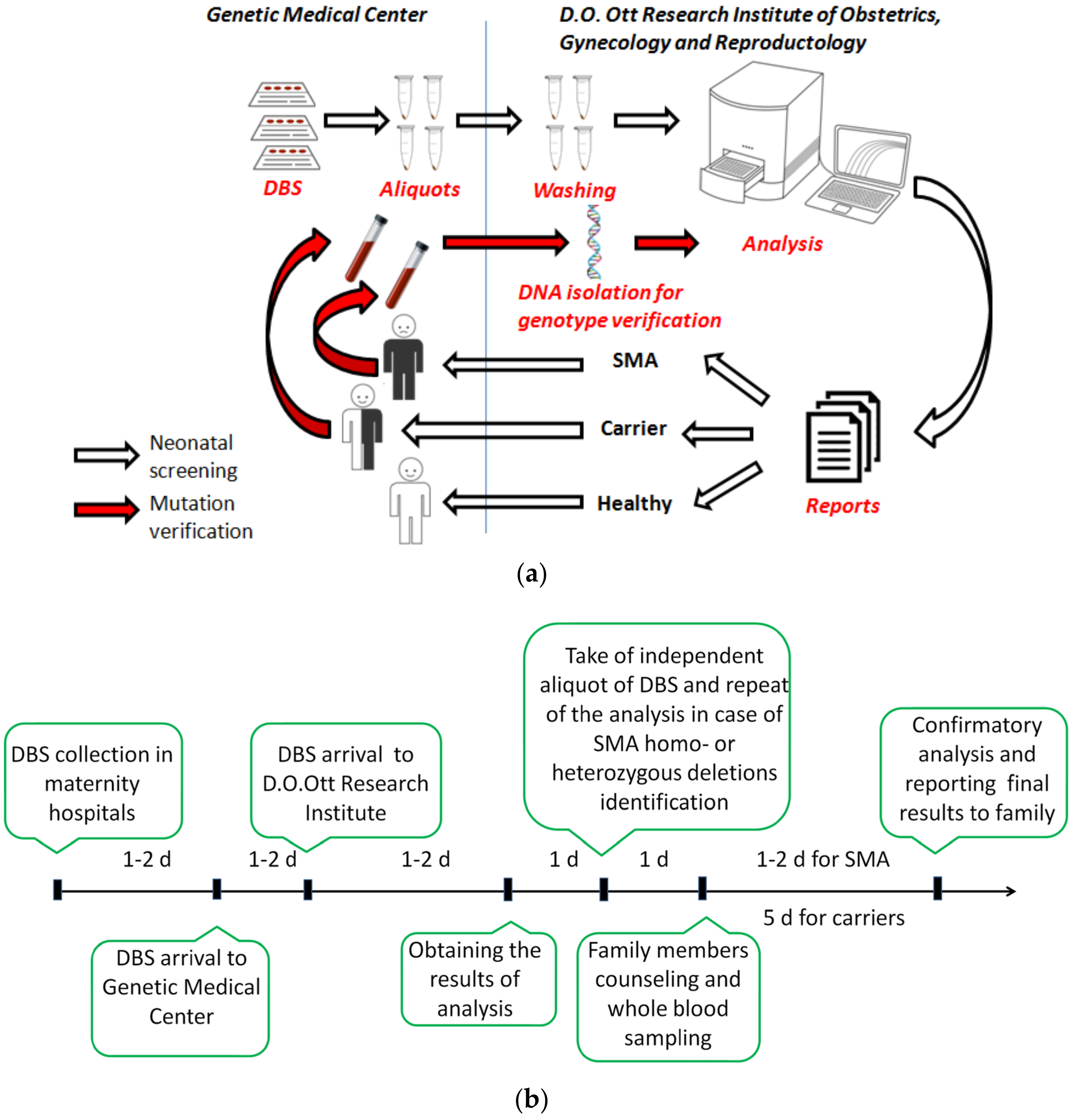
2. Materials and Methods
2.1. Study Recruitment
2.2. Assay Validation
2.3. Specimens
2.4. SMA Screening Assay
2.4.1. DNA Extraction
- A 1× GenomeX elution reagent was prepared by diluting the 100× solution with ampouled or bidistilled water 1:99 to obtain 250 µL of 1× solution per sample.
- A 3.2 mm diameter circle was punched out from a DBS card into a 0.2 mL tube or a 96-well plate using a puncher.
- 100 µL of 1× elution reagent was added to each tube.
- Next, the tubes were incubated for 15 min at room temperature.
- The liquid was stirred by pipetting twice and then removed from the tube.
- Steps 3–5 were repeated one more time.
- 50 mL of 1× elution reagent was added to the washed filter.
- The tubes/plated were closed and centrifuged to remove droplets from the lids.
- The samples were heated for 15 min at +99 °C in a hot-lid Bio-Rad T100 thermocycler.
- The samples were removed to +4 °C and stored at that temperature for up to 3 weeks.
2.4.2. Performing Real-Time PCR
- GenomeX MasterMix was defrosted and added to the sample, along with polymerase (17.6 µL of MasterMix + 0.4 µL of polymerase per sample). Each DNA sample was analyzed twice.
- 18 µL of prepared mix was added to each well of a 96-well plate.
- 2 µL of DNA sample was added to each well. No DNA was added to 2 of the wells (the contamination controls).
- The plate was centrifuged to remove droplets from the lid.
- The plate was placed in a real-time PCR device under the following conditions: preincubation, 95 °C for 5 min, then 40 cycles of 94 °C for 15 s and 62 °C for 1 min.
2.4.3. Analysis of the Results
2.5. Screening, Notification of Results and Confirmatory Testing
3. Results
3.1. Validation Study
3.2. Flow of Patient Samples and Data
3.3. Screening for SMA
3.4. Determination of the of Frequency SMA Carrier Status in Newborns
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ogino, S.; Wilson, R.B. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum. Genet. 2002, 111, 477–500. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, J.A.; Singh, P.; Darras, B.T. Spinal muscular atrophy: A clinical and research update. Pediatr. Neurol. 2012, 46, 1–12. [Google Scholar] [CrossRef]
- Russman, B.S. Spinal muscular atrophy: Clinical classification and disease heterogeneity. J. Child Neurol. 2007, 22, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Zerres, K.; Wirth, B.; Rudnik-Schöneborn, S. Spinal muscular atrophy—Clinical and genetic correlations. Neuromuscul. Disord. 1997, 7, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Glotov, A.S.; Kiselev, A.V.; Ivashchenko, T.E.; Baranov, V.S. Analysis of deletional damage in SMN1, SMN2, and NAIP genes in patients with spinal muscular atrophy in the northwestern region of Russia. Genetika 2001, 37, 1156–1159. [Google Scholar] [PubMed]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.M.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Cuscó, I.; Barceló, M.J.; Rojas–García, R.; Illa, I.; Gámez, J.; Cervera, C.; Pou, A.; Izquierdo, G.; Baiget, M.; Tizzano, E.F. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J. Neurol. 2006, 253, 21–25. [Google Scholar] [CrossRef]
- Zheleznyakova, G.Y.; Nilsson, E.K.; Kiselev, A.V.; Maretina, M.A.; Tishchenko, L.I.; Fredriksson, R.; Baranov, V.S.; Schiöth, H.B. Methylation levels of SLC23A2 and NCOR2 genes correlate with spinal muscular atrophy severity. PLoS ONE 2015, 10, e0121964. [Google Scholar] [CrossRef] [PubMed]
- Hosseinibarkooie, S.; Schneider, S.; Wirth, B. Advances in understanding the role of disease-associated proteins in spinal muscular atrophy. Exp. Rev. Proteom. 2017, 14, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Maretina, M.A.; Zheleznyakova, G.Y.; Lanko, K.M.; Egorova, A.A.; Baranov, V.S.; Kiselev, A.V. Molecular Factors Involved in Spinal Muscular Atrophy Pathways as Possible Disease-modifying Candidates. Curr. Genom. 2018, 19, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Maretina, M.; Egorova, A.; Baranov, V.; Kiselev, A. DYNC1H1 gene methylation correlates with severity of spinal muscular atrophy. Ann. Hum. Genet. 2019, 83, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Zheleznyakova, G.Y.; Kiselev, A.V.; Vakharlovsky, V.G.; Rask-Andersen, M.; Chavan, R.; Egorova, A.A.; Schiöth, H.B.; Baranov, V.S. Genetic and expression studies of SMN2 gene in Russian patients with spinal muscular atrophy type II and III. BMC Med. Genet. 2011, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Bernal, S.; Also-Rallo, E.; Martínez-Hernández, R.; Alías, L.; Rodríguez-Alvarez, F.J.; Millán, J.M.; Hernández-Chico, C.; Baiget, M.; Tizzano, E.F. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul. Disord. 2011, 21, 413–419. [Google Scholar] [CrossRef]
- Rigo, F.; Hua, Y.; Krainer, A.R.; Bennett, C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012, 199, 21–25. [Google Scholar] [CrossRef]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef]
- Poirier, A.; Weetall, M.; Heinig, K.; Bucheli, F.; Schoenlein, K.; Alsenz, J.; Bassett, S.; Ullah, M.; Senn, C.; Ratni, H.; et al. Risdiplam distributes and increases <scp>SMN</scp> protein in both the central nervous system and peripheral organs. Pharmacol. Res. Perspect. 2018, 6, e00447. [Google Scholar] [CrossRef]
- Glascock, J.J.; Osman, E.Y.; Wetz, M.J.; Krogman, M.M.; Shababi, M.; Lorson, C.L. Decreasing Disease Severity in Symptomatic, Smn −/−; SMN2 +/+, Spinal Muscular Atrophy Mice Following scAAV9-SMN Delivery. Hum. Gene Ther. 2012, 23, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Benkhelifa-Ziyyat, S.; Besse, A.; Roda, M.; Duque, S.; Astord, S.; Carcenac, R.; Marais, T.; Barkats, M. Intramuscular scAAV9-SMN Injection Mediates Widespread Gene Delivery to the Spinal Cord and Decreases Disease Severity in SMA Mice. Mol. Ther. 2013, 21, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Faravelli, I.; Meneri, M.; Saccomanno, D.; Velardo, D.; Abati, E.; Gagliardi, D.; Parente, V.; Petrozzi, L.; Ronchi, D.; Stocchetti, N.; et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on Spinal Muscular Atrophy type 3 patients. J. Cell Mol. Med. 2020, 24, 3034–3039. [Google Scholar] [CrossRef] [PubMed]
- Govoni, A.; Gagliardi, D.; Comi, G.P.; Corti, S. Time Is Motor Neuron: Therapeutic Window and Its Correlation with Pathogenetic Mechanisms in Spinal Muscular Atrophy. Mol. Neurobiol. 2018, 55, 6307–6318. [Google Scholar] [CrossRef]
- Anhuf, D.; Eggermann, T.; Rudnik-Schöneborn, S.; Zerres, K. Determination of SMN1 and SMN2 copy number using TaqManTM technology. Hum. Mutat. 2003, 22, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Aljanabi, S. Universal and rapid salt-extraction of high quality genomic DNA for PCR- based techniques. Nucleic Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy—A literature review. Orphanet. J. Rare Dis. 2017, 12, 124. [Google Scholar] [CrossRef]
- Kraszewski, J.N.; Kay, D.M.; Stevens, C.F.; Koval, C.; Haser, B.; Ortiz, V.; Albertorio, A.; Cohen, L.L.; Jain, R.; Andrew, S.P.; et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet. Med. 2018, 20, 608–613. [Google Scholar] [CrossRef]
- Gailite, L.; Sterna, O.; Konika, M.; Isakovs, A.; Isakova, J.; Micule, I.; Setlere, S.; Diriks, M.; Auzenbaha, M. New-Born Screening for Spinal Muscular Atrophy: Results of a Latvian Pilot Study. Int. J. Neonatal Screen. 2022, 8, 15. [Google Scholar] [CrossRef]
- Scarciolla, O.; Stuppia, L.; De Angelis, M.V.; Murru, S.; Palka, C.; Giuliani, R.; Pace, M.; Di Muzio, A.; Torrente, I.; Morella, A.; et al. Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics 2006, 7, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Dangouloff, T.; Vrščaj, E.; Servais, L.; Osredkar, D.; Adoukonou, T.; Aryani, O.; Barisic, N.; Bashiri, F.; Bastaki, L.; Benitto, A.; et al. Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go. Neuromuscul. Disord. 2021, 31, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Zabnenkova, V.V.; Dadali, E.L.; Spiridonova, M.G.; Zinchenko, R.A.; Polyakov, A. V Heterozygous carrier rate for type I–IV proximal spinal muscular atrophy in Chuvashes, Udmurts, and residents of the Moscow region. Russ. J. Genet. 2012, 48, 838–845. [Google Scholar] [CrossRef]
- Lin, Y.; Lin, C.-H.; Yin, X.; Zhu, L.; Yang, J.; Shen, Y.; Yang, C.; Chen, X.; Hu, H.; Ma, Q.; et al. Newborn Screening for Spinal Muscular Atrophy in China Using DNA Mass Spectrometry. Front. Genet. 2019, 10, 1255. [Google Scholar] [CrossRef] [PubMed]
- Mikhalchuk, K.; Shchagina, O.; Chukhrova, A.; Zabnenkova, V.; Chausova, P.; Ryadninskaya, N.; Vlodavets, D.; Kutsev, S.I.; Polyakov, A. Pilot Program of Newborn Screening for 5q Spinal Muscular Atrophy in the Russian Federation. Int. J. Neonatal Screen. 2023, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Vill, K.; Schwartz, O.; Blaschek, A.; Gläser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Röschinger, W.; Becker, M.; Czibere, L.; et al. Newborn screening for spinal muscular atrophy in Germany: Clinical results after 2 years. Orphanet. J. Rare Dis. 2021, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Abiusi, E.; Vaisfeld, A.; Fiori, S.; Novelli, A.; Spartano, S.; Faggiano, M.V.; Giovanniello, T.; Angeloni, A.; Vento, G.; Santoloci, R.; et al. Experience of a 2-year spinal muscular atrophy NBS pilot study in Italy: Towards specific guidelines and standard operating procedures for the molecular diagnosis. J. Med. Genet. 2023, 60, 697–705. [Google Scholar] [CrossRef]
- Chien, Y.H.; Chiang, S.C.; Weng, W.C.; Lee, N.C.; Lin, C.J.; Hsieh, W.S.; Lee, W.T.; Jong, Y.J.; Ko, T.M.; Hwu, W.L. Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening. J. Pediatr. 2017, 190, 124–129.e1. [Google Scholar] [CrossRef]
- Hale, K.; Ojodu, J.; Singh, S. Landscape of spinal muscular atrophy newborn screening in the united states: 2018–2021. Int. J. Neonatal Screen. 2021, 7, 33. [Google Scholar] [CrossRef]
- Boemer, F.; Caberg, J.-H.; Beckers, P.; Dideberg, V.; di Fiore, S.; Bours, V.; Marie, S.; Dewulf, J.; Marcelis, L.; Deconinck, N.; et al. Three years pilot of spinal muscular atrophy newborn screening turned into official program in Southern Belgium. Sci. Rep. 2021, 11, 19922. [Google Scholar] [CrossRef]
- Kariyawasam, D.S.T.; Russell, J.S.; Wiley, V.; Alexander, I.E.; Farrar, M.A. The implementation of newborn screening for spinal muscular atrophy: The Australian experience. Genet. Med. 2020, 22, 557–565. [Google Scholar] [CrossRef]
- Yeh, E.; Amburgey, K.; Boyd, J.; Campbell, C.; Dowling, J.J.; Gonorazky, H.; Marcadier, J.; Tarnopolsky, M.A.; Vajsar, J.; MacKenzie, A.; et al. Newborn Screening for Spinal Muscular Atrophy: Ontario Testing and Follow-up Recommendations. Can. J. Neurol. Sci. 2021, 48, 504–511. [Google Scholar] [CrossRef]
- Smith, M.; Calabro, V.; Chong, B.; Gardiner, N.; Cowie, S.; du Sart, D. Population screening and cascade testing for carriers of SMA. Eur. J. Hum. Genet. 2007, 15, 759–766. [Google Scholar] [CrossRef]
- Li, S.; Han, X.; Xu, Y.; Chang, C.; Gao, L.; Li, J.; Lu, Y.; Mao, A.; Wang, Y. Comprehensive Analysis of Spinal Muscular Atrophy. J. Mol. Diagn. 2022, 24, 1009–1020. [Google Scholar] [CrossRef]
- Shih, S.T.F.; Farrar, M.A.; Wiley, V.; Chambers, G. Newborn screening for spinal muscular atrophy with disease-modifying therapies: A cost-effectiveness analysis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1296–1304. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Genotype | Wt/Wt | Del 7/Wt | Del 7/Del 7 | Sensitivity */ Specificity |
|---|---|---|---|---|
| Del 7/Del 7 | 0 | 0 | 50 | 100%/100% |
| Del 7/Wt | 0 | 58 | 0 | 100%/100% |
| Wt/Wt | 142 | 0 | 0 | 100%/100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiselev, A.; Maretina, M.; Shtykalova, S.; Al-Hilal, H.; Maslyanyuk, N.; Plokhih, M.; Serebryakova, E.; Frolova, M.; Shved, N.; Krylova, N.; et al. Establishment of a Pilot Newborn Screening Program for Spinal Muscular Atrophy in Saint Petersburg. Int. J. Neonatal Screen. 2024, 10, 9. https://doi.org/10.3390/ijns10010009
Kiselev A, Maretina M, Shtykalova S, Al-Hilal H, Maslyanyuk N, Plokhih M, Serebryakova E, Frolova M, Shved N, Krylova N, et al. Establishment of a Pilot Newborn Screening Program for Spinal Muscular Atrophy in Saint Petersburg. International Journal of Neonatal Screening. 2024; 10(1):9. https://doi.org/10.3390/ijns10010009
Chicago/Turabian StyleKiselev, Anton, Marianna Maretina, Sofia Shtykalova, Haya Al-Hilal, Natalia Maslyanyuk, Mariya Plokhih, Elena Serebryakova, Marina Frolova, Natalia Shved, Nadezhda Krylova, and et al. 2024. "Establishment of a Pilot Newborn Screening Program for Spinal Muscular Atrophy in Saint Petersburg" International Journal of Neonatal Screening 10, no. 1: 9. https://doi.org/10.3390/ijns10010009
APA StyleKiselev, A., Maretina, M., Shtykalova, S., Al-Hilal, H., Maslyanyuk, N., Plokhih, M., Serebryakova, E., Frolova, M., Shved, N., Krylova, N., Il’ina, A., Freund, S., Osinovskaya, N., Sultanov, I., Egorova, A., Lobenskaya, A., Koroteev, A., Sosnina, I., Gorelik, Y., ... Glotov, A. (2024). Establishment of a Pilot Newborn Screening Program for Spinal Muscular Atrophy in Saint Petersburg. International Journal of Neonatal Screening, 10(1), 9. https://doi.org/10.3390/ijns10010009