
Neonatal Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency—Alternative Approaches

{kind=link}
Abstract
: The challenges inherent in many newborn screening programmes for medium-chain acyl-CoA dehydrogenase deficiency could be overcome by better use of the available second-line tests. Application of “next generation” technologies could minimize many of the problems generated by the current approach to genetic analysis. (Comment on Maier EM. Int. J. Neonatal Screen. 2015, 1, 79–88)To the Editor
Conflicts of Interest
Abbreviations
| MCAD | Medium-chain acyl-CoA dehydrogenase |
| MCADD | Medium-chain acyl-CoA dehydrogenase deficiency |
References
- Oerton, J.; Khalid, J.M.; Besley, G.; Dalton, R.N.; Downing, M.; Green, A.; Henderson, M.; Krywawych, S.; Leonard, J.; Andresen, B.S.; et al. Newborn screening for medium chain acyl-CoA dehydrogenase deficiency in England: Prevalence, predictive value and test validity based on 1.5 million screened babies. J. Med. Screen. 2011, 4, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Andresen, B.S.; Pedersen, C.B.; Olsen, R.K.J.; Corydon, T.J.; Bross, P. Mitochondrial fatty acid oxidation defects—remaining challenges. J. Inherit. Metab. Dis. 2008, 31, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Derks, T.G.; Boer, T.S.; van Assen, A.; Bos, T.; Ruiter, J.; Waterham, H.R.; Niezen-Koning, K.E.; Wanders, R.J.A.; Rondeel, J.M.M.; Loeber, J.; et al. Neonatal screening for medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in The Netherlands: The importance of enzyme analysis to ascertain true MCAD deficiency. J. Inherit. Metab. Dis. 2008, 31, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Gramar, G.; Haege, G.; Fang-Hoffmann, J.; Hoffmann, G.F.; Bartram, C.R.; Hinderhofer, K.; Burgard, P.; Lindner, M. Medium-chain acyl-CoA dehydrogenase deficiency: Evaluation of genotype-phenotype correlation in patients detected by newborn screening. JIMD Rep. 2015, 101–112. [Google Scholar]
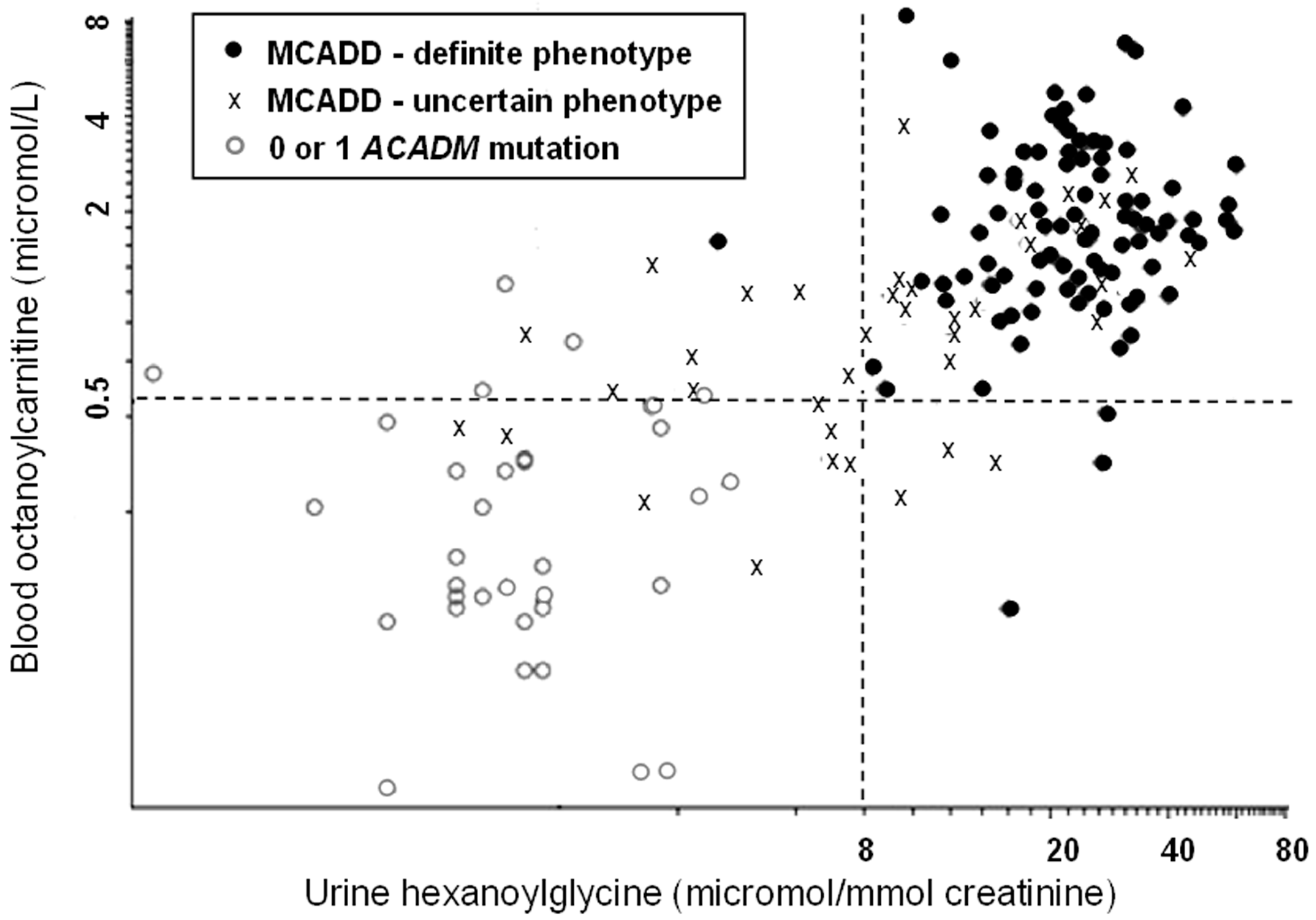
- Downing, M.; Manning, N.J.; Dalton, R.N.; Krywawych, S.; Oerton, J. Detection of urinary hexanoylglycine in the diagnosis of MCAD deficiency from newborn screening. J. Inherit. Metab. Dis. 2008, 31, 550. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pollitt, R.J. Neonatal Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency—Alternative Approaches. Int. J. Neonatal Screen. 2016, 2, 1. https://doi.org/10.3390/ijns2010001
Pollitt RJ. Neonatal Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency—Alternative Approaches. International Journal of Neonatal Screening. 2016; 2(1):1. https://doi.org/10.3390/ijns2010001
Chicago/Turabian StylePollitt, Rodney J. 2016. "Neonatal Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency—Alternative Approaches" International Journal of Neonatal Screening 2, no. 1: 1. https://doi.org/10.3390/ijns2010001
APA StylePollitt, R. J. (2016). Neonatal Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency—Alternative Approaches. International Journal of Neonatal Screening, 2(1), 1. https://doi.org/10.3390/ijns2010001