Current Practices for U.S. Newborn Screening of Pompe Disease and MPSI


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
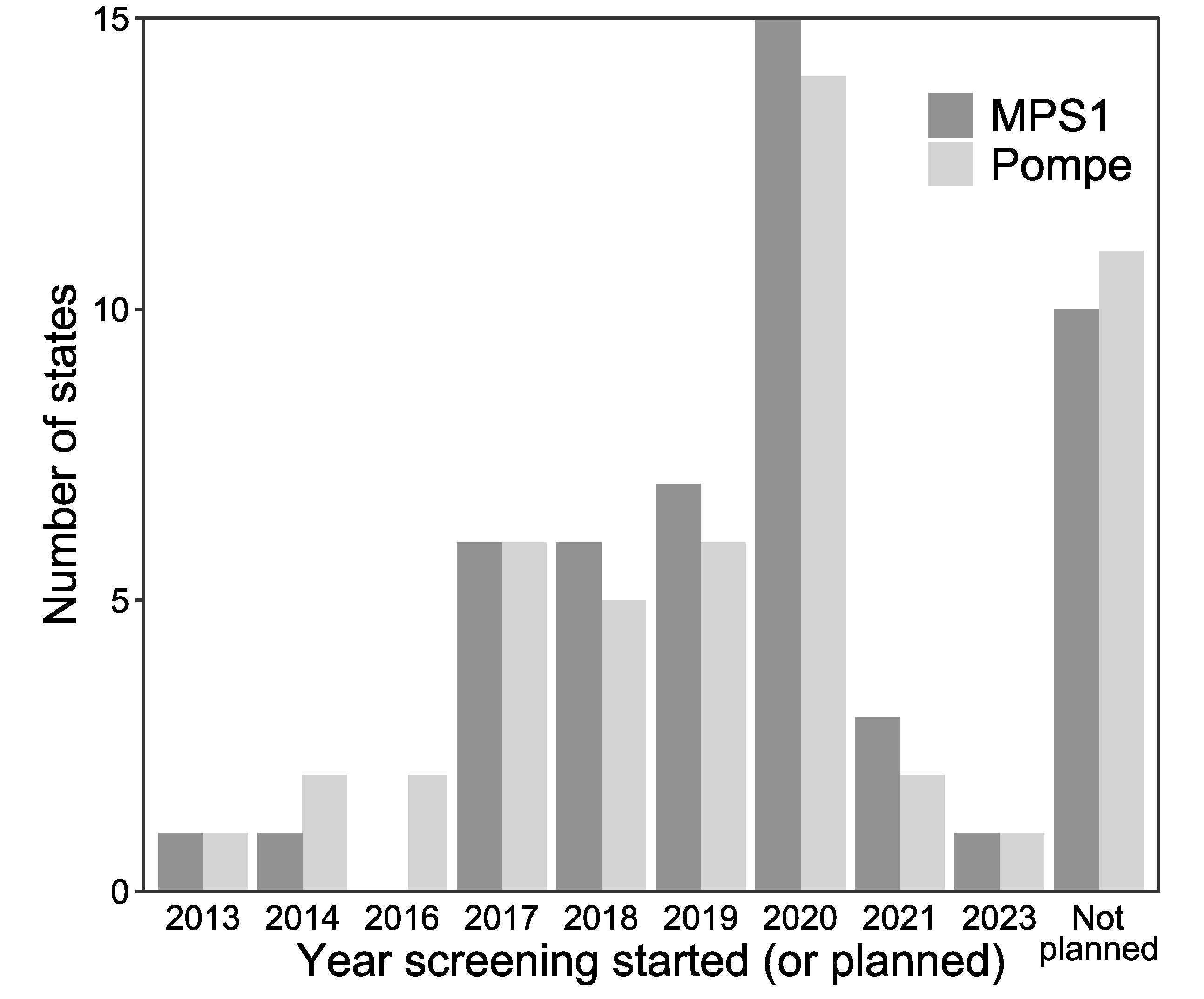
3. Results
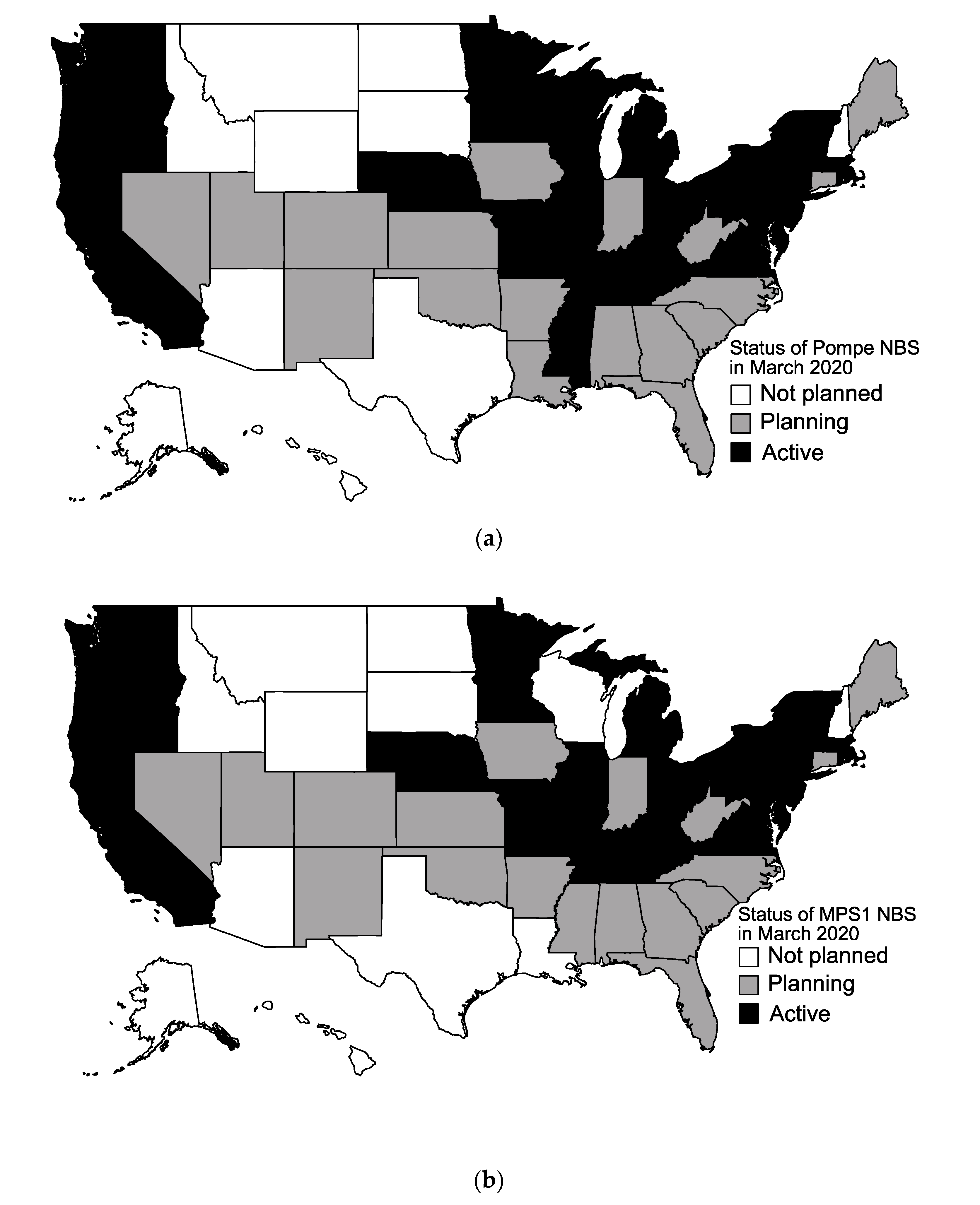
3.1. Current State of Newborn Screening for Lysosomal Storage Diseases in the U.S.
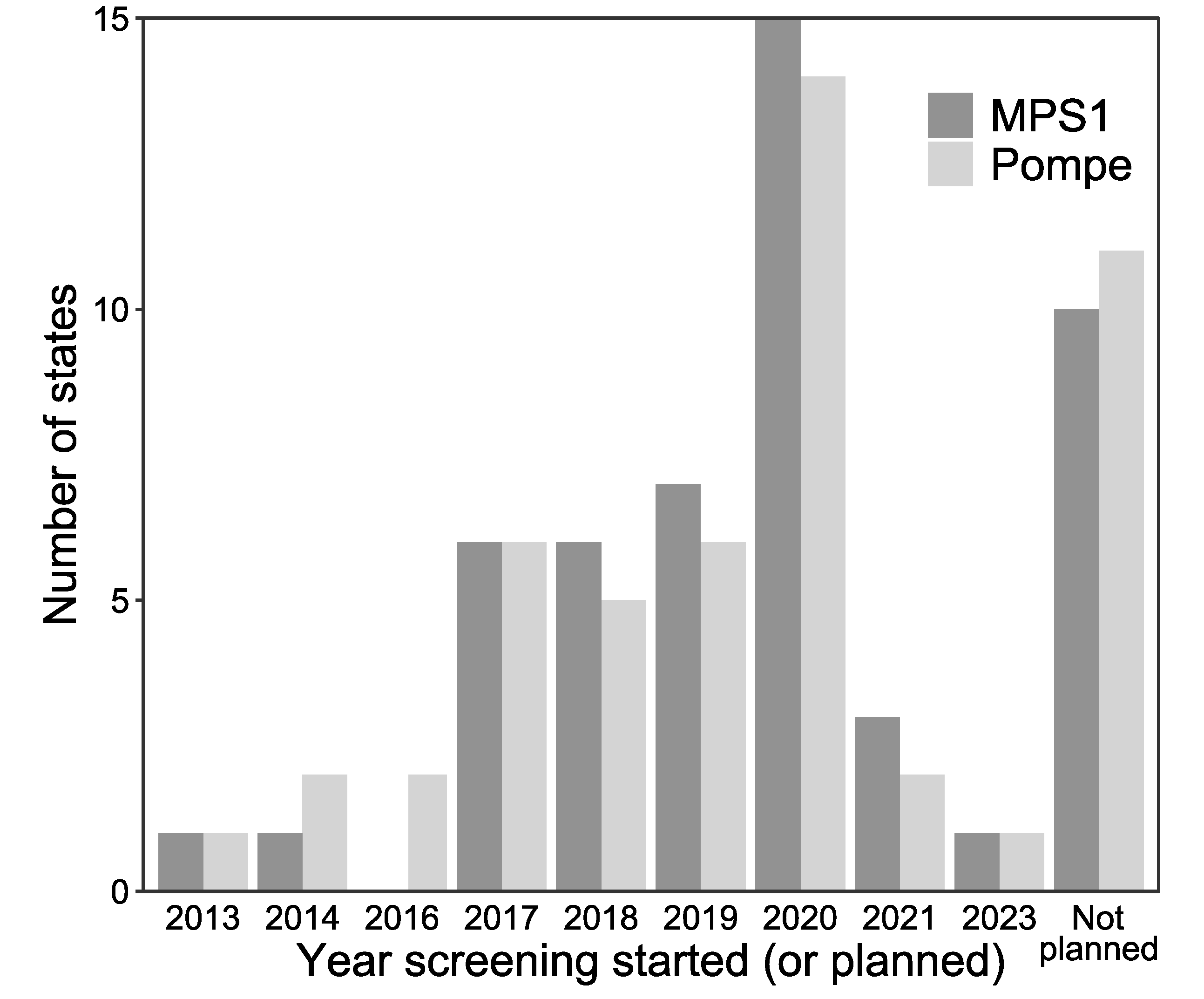
3.2. LSD NBS Planned Expansion by the End of 2020
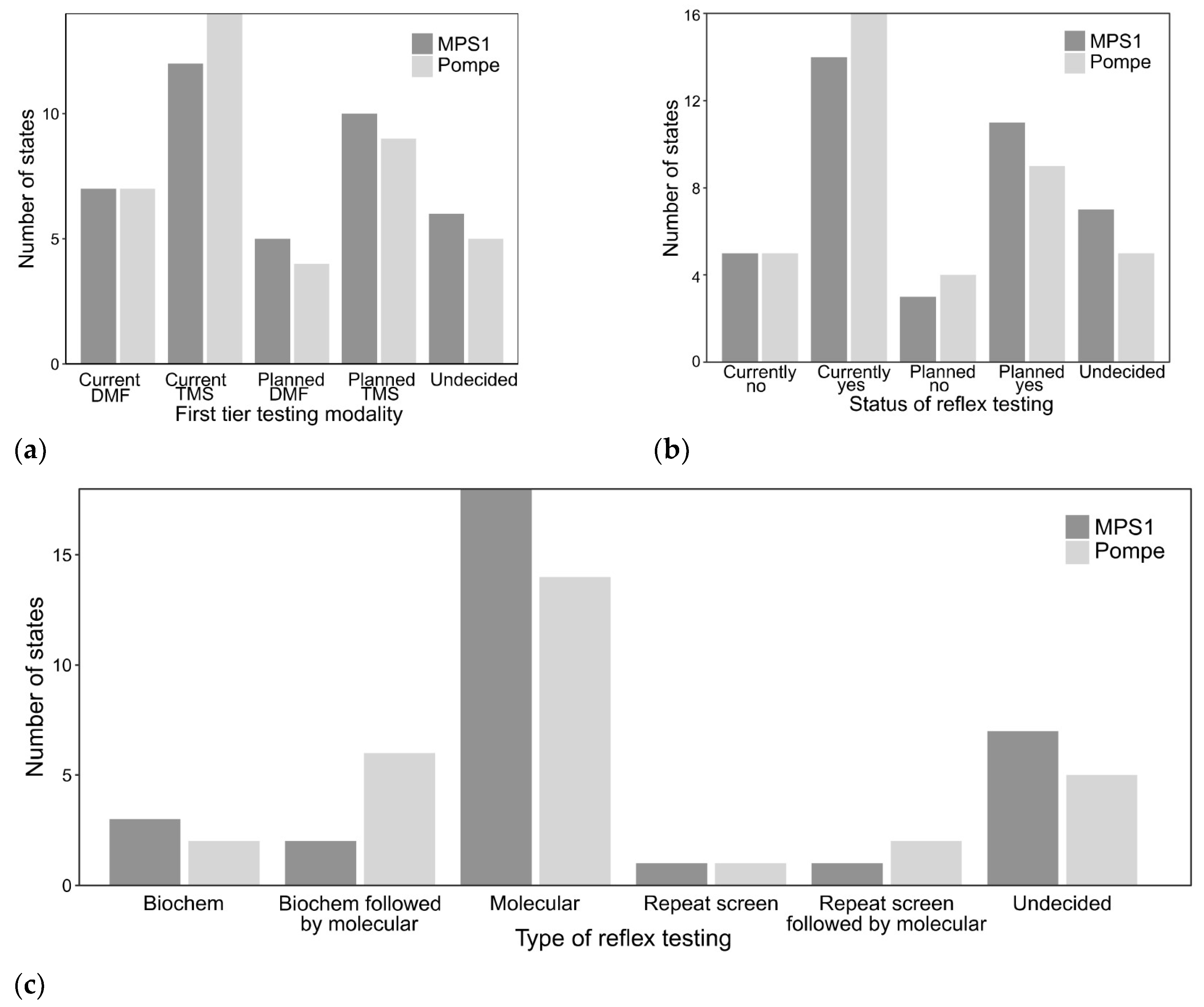
3.3. LSD NBS by Analyte and Use of Reflex Testing
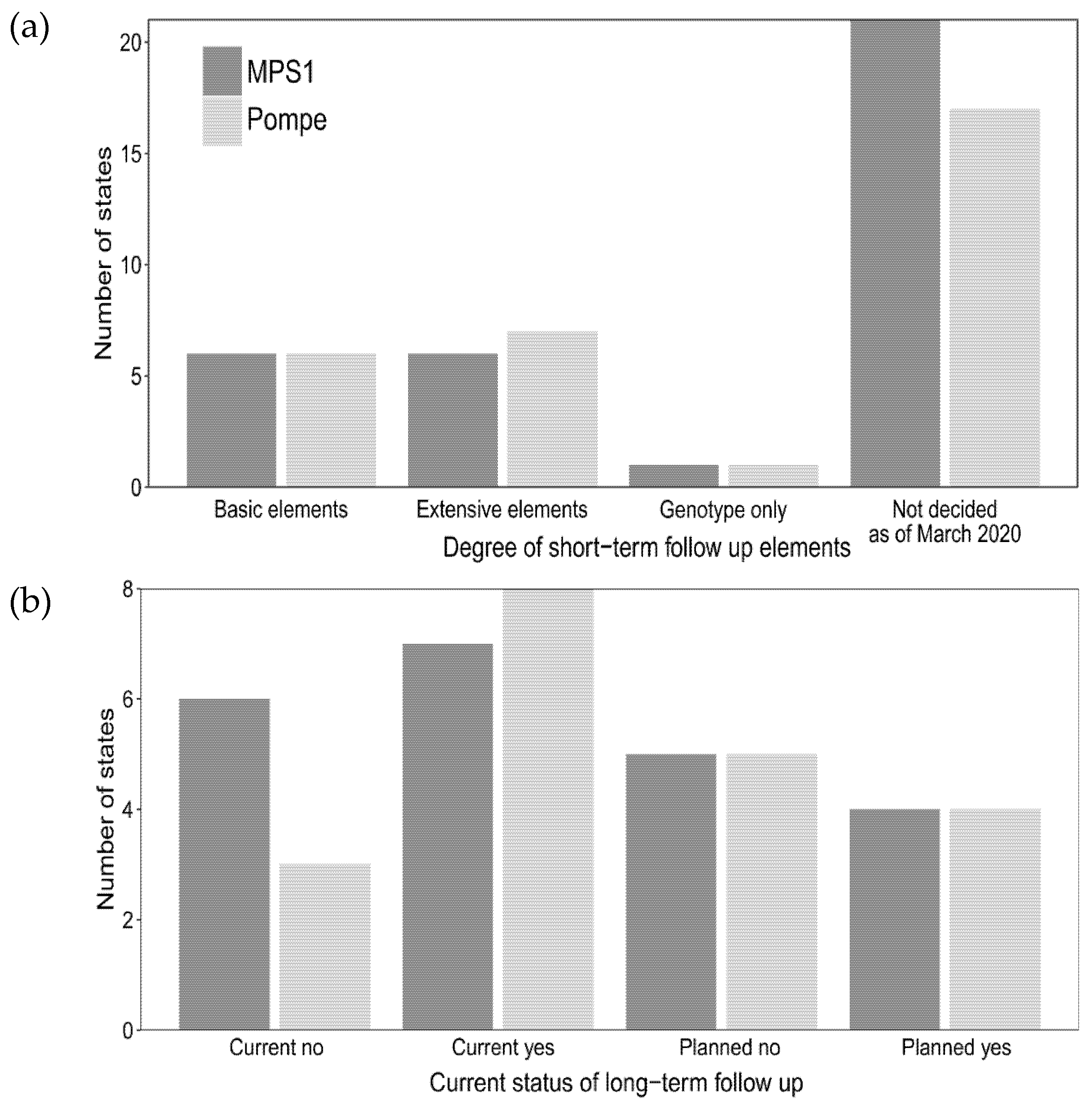
3.4. Practices for Both Short-and Long-Term Follow-Up of Newborn Screening
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Executive Summary of Adding Pompe to RUSP. 2015. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/rusp/previous-nominations/pompe-exsum.pdf (accessed on 1 June 2020).
- Executive Summary of Adding MPSI to RUSP. 2016. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/rusp/previous-nominations/mps-i-exsum.pdf (accessed on 1 June 2020).
- Eisengart, J.B.; Rudser, K.D.; Xue, Y.; Orchard, P.; Miller, W.; Lund, T.; Van Der Ploeg, A.; Mercer, J.; Jones, S.; Mengel, K.E.; et al. Long-term outcomes of systemic therapies for Hurler syndrome: An international multicenter comparison. Genet. Med. 2018, 20, 1423–1429. [Google Scholar] [CrossRef] [Green Version]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.F.; Yang, C.C.; Liao, H.C.; Huang, L.Y.; Chiang, C.C.; Ho, H.C.; Lai, C.-J.; Chu, T.-H.; Yang, T.-F.; Hsu, T.-R.; et al. Very Early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes. J. Pediatr. 2016, 169, 174–180. [Google Scholar] [CrossRef]
- Yang, C.F.; Liu, H.C.; Hsu, T.R.; Tsai, F.C.; Chiang, S.F.; Chiang, C.C.; Ho, H.-C.; Lai, C.-J.; Yang, T.-F.; Chuang, S.-Y.; et al. A Large-Scale Nationwide Newborn Screening Program for Pompe Disease in Taiwan: Towards Effective Diagnosis and Treatment. Am. J. Med. Genet. A 2014, 164, 54–61. [Google Scholar]
- Kishnani, P.S.; Corzo, D.; Nicolino, M.; Byrne, B.; Mandel, H.; Hwu, W.; Leslie, N.; Levine, J.; Spencer, C.; McDonald, M.; et al. Recombinant human acid -glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology 2006, 68, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Labrousse, P.; Chien, Y.H.; Pomponio, R.J.; Keutzer, J.; Lee, N.C.; Akmaev, V.R.; Scholl, T.; Hwu, W. Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. Mol. Genet. Metab. 2010, 99, 379–383. [Google Scholar] [CrossRef]
- Taylor, J.L.; Clinard, K.; Powell, C.M.; Rehder, C.; Young, S.P.; Bali, D.; Beckloff, S.E.; Gehtland, L.M.; Kemper, A.R.; Lee, S.; et al. The North Carolina Experience with Mucopolysaccharidosis Type I Newborn Screening. J. Pediatr. 2019, 211, 193–200. [Google Scholar] [CrossRef]
- Orsini, J.J.; The New York State Krabbe Disease Consortium; Kay, D.M.; Saavedra-Matiz, C.A.; Wenger, D.A.; Duffner, P.K.; Erbe, R.W.; Biski, C.; Martin, M.; Krein, L.M.; et al. Newborn screening for Krabbe disease in New York State: The first eight years’ experience. Genet. Med. 2016, 18, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision newborn screening for lysosomal disorders. Genet. Med. 2018, 20, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Aronovich, E.L.; Pan, D.; Whitley, C.B. Molecular Genetic Defect Underlying Alpha-L-Iduronidase Pseudodeficiency. Am. J. Hum. Genet. 1996, 58, 75–85. [Google Scholar]
- Filocamo, M.; Tomanin, R.; Bertola, F.; Morrone, A. Biochemical and Molecular Analysis in Mucopolysaccharidoses: What a Paediatrician Must Know. Ital. J. Pediatr. 2018, 44, 129. [Google Scholar]
- Chien, Y.H.; Tsai, W.H.; Chang, C.L.; Chiu, P.C.; Chou, Y.Y.; Tsai, F.J.; Wong, S.-L.; Lee, N.-C.; Hwu, W. Earlier and Higher Dosing of Alglucosidase Alfa Improve Outcomes in Patients with Infantile-Onset Pompe Disease: Evidence from Real-World Experiences. Mol. Genet. Metab. Rep. 2020, 23, 100591. [Google Scholar]
- Grosse, S.D.; Lam, W.K.K.; Wiggins, L.D.; Kemper, A.R. Cognitive outcomes and age of detection of severe mucopolysaccharidosis type 1. Genet. Med. 2017, 19, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Momosaki, K.; Kido, J.; Yoshida, S.; Sugawara, K.; Miyamoto, T.; Inoue, T.; Okumiya, T.; Matsumoto, S.; Endo, F.; Hirose, S.; et al. Newborn screening for Pompe disease in Japan: Report and literature review of mutations in the GAA gene in Japanese and Asian patients. J. Hum. Genet. 2019, 64, 741–755. [Google Scholar] [CrossRef]
- Clarke, L.A.; Atherton, A.M.; Burton, B.K.; Day-Salvatore, D.L.; Kaplan, P.; Leslie, N.D.; Scott, C.R.; Stockton, D.W.; Thomas, J.A.; Muenzer, J. Mucopolysaccharidosis Type I Newborn Screening: Best Practices for Diagnosis and Management. J. Pediatr. 2017, 182, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Lee, C.L.; Chuang, C.K.; Lin, S.P. Long-Term Outcomes of Enzyme Replacement Therapy for Taiwanese Patients with Mucopolysaccharidosis. I. Pediatr. Neonatol 2019, 60, 577–578. [Google Scholar]
- Wickham, H.; Chang, W.; Henry, L.; Pedersen, T.; Takahashi, K.; Wilke, C. Create Elegant Data Visualisations Using the Grammar of Graphics R Package Version 3.5.1. 2019. Available online: https://cran.r-project.org/package=ggplot2 (accessed on 1 June 2020).
- Tortorelli, S.; Eckerman, J.S.; Orsini, J.J.; Stevens, C.; Hart, J.; Hall, P.L.; Alexander, J.J.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; et al. Moonlighting newborn screening markers: The incidental discovery of a second-tier test for Pompe disease. Genet. Med. 2018, 20, 840–846. [Google Scholar]
- Peck, D.S.; Lacey, J.M.; White, A.L.; Pino, G.; Studinski, A.L.; Fisher, R.; Ahmad, A.; Spencer, L.; Viall, S.; Shallow, N.; et al. Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I. Int. J. Neonatal Screen. 2020, 6, 10. [Google Scholar]
- Matern, D.; Tortorelli, S.; Oglesbee, D.; Gavrilov, D.; Rinaldo, P. Reduction of the False-Positive Rate in Newborn Screening by Implementation of MS/MS-Based Second-Tier Tests: The Mayo Clinic Experience (2004–2007). Int. J. Inherit. Metab. Dis. 2007, 30, 585–592. [Google Scholar]
- Turgeon, C.T.; Magera, M.J.; Cuthbert, C.D.; Loken, P.R.; Gavrilov, D.K.; Tortorelli, S.; Raymond, K.M.; Oglesbee, D.; Rinaldo, P.; Matern, D. Determination of Total Homocysteine, Methylmalonic Acid, and 2-Methylcitric Acid in Dried Blood Spots by Tandem Mass Spectrometry. Clin. Chem. 2010, 56, 1686–1695. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, C.T.; Orsini, J.J.; Sanders, K.A.; Magera, M.J.; Langan, T.J.; Escolar, M.L.; Duffner, P.; Oglesbee, D.; Gavrilov, D.; Tortorelli, S.; et al. Measurement of psychosine in dried blood spots—A possible improvement to newborn screening programs for Krabbe disease. J. Inherit. Metab. Dis. 2015, 38, 923–929. [Google Scholar] [CrossRef]
- de Ruijter, J.; de Ru, M.H.; Wagemans, T.; IJlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; Van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef]
- Peng, G.; Shen, P.; Gandotra, N.; Le, A.; Fung, E.; Jelliffe-Pawlowski, L.; Davis, R.W.; Enns, G.M.; Zhao, H.; Cowan, T.M.; et al. Combining Newborn Metabolic and DNA Analysis for Second-Tier Testing of Methylmalonic Acidemia. Genet. Med. 2019, 21, 896–903. [Google Scholar]
- Smith, L.D.; Bainbridge, M.N.; Parad, R.B.; Bhattacharjee, A. Second Tier Molecular Genetic Testing in Newborn Screening for Pompe Disease: Landscape and Challenges. Int. J. Neonatal Screen. 2020, 6, 32. [Google Scholar]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal. Screen. 2019, 5, 24. [Google Scholar]
- Polo, G.; Gueraldi, D.; Giuliani, A.; Rubert, L.; Cazzorla, C.; Salviati, L.; Marzollo, A.; Biffi, A.; Burlina, A.P.; Burlina, A.B. The Combined Use of Enzyme Activity and Metabolite Assays as a Strategy for Newborn Screening of Mucopolysaccharidosis Type I. Clin. Chem. Lab. Med. 2020. [Google Scholar] [CrossRef]
- Scarpa, M.; Almássy, Z.; Beck, M.; Bodamer, O.; Bruce, I.A.; De Meirleir, L.; Guffon, N.; Guillén-Navarro, E.; Hensman, P.; Jones, S.A.; et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J. Rare Dis. 2011, 6, 72. [Google Scholar]
- Morrison, D.R.; Clayton, E.W. False positive newborn screening results are not always benign. Public Heal. Genom. 2011, 14, 173–177. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Albers, S.; Amato, S.; Ampola, M.; Brewster, T.G.; Demmer, L.; Eaton, R.B.; Greenstein, R.; Korson, M.; Larson, C.; et al. Effect of Expanded Newborn Screening for Biochemical Genetic Disorders on Child Outcomes and Parental Stress. JAMA 2003, 290, 2564–2572. [Google Scholar]
- Wasserstein, M.P. Long-term follow-up in newborn screening: The role of collaboration. Genet. Med. 2016. [Google Scholar] [CrossRef] [Green Version]
- Hinton, C.F.; Homer, C.J.; Thompson, A.A.; Williams, A.; Hassell, K.L.; Feuchtbaum, L.; Berry, S.A.; Comeau, A.M.; Therrell, B.L.; Brower, A.; et al. A framework for assessing outcomes from newborn screening: On the road to measuring its promise. Mol. Genet. Metab. 2016, 118, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Berry, S.A.; Lloyd-Puryear, M.A.; Watson, M.S. Long-term follow-up of newborn screening patients. Genet. Med. 2010, 12. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ames, E.G.; Fisher, R.; Kleyn, M.; Ahmad, A. Current Practices for U.S. Newborn Screening of Pompe Disease and MPSI. Int. J. Neonatal Screen. 2020, 6, 72. https://doi.org/10.3390/ijns6030072
Ames EG, Fisher R, Kleyn M, Ahmad A. Current Practices for U.S. Newborn Screening of Pompe Disease and MPSI. International Journal of Neonatal Screening. 2020; 6(3):72. https://doi.org/10.3390/ijns6030072
Chicago/Turabian StyleAmes, Elizabeth G., Rachel Fisher, Mary Kleyn, and Ayesha Ahmad. 2020. "Current Practices for U.S. Newborn Screening of Pompe Disease and MPSI" International Journal of Neonatal Screening 6, no. 3: 72. https://doi.org/10.3390/ijns6030072
APA StyleAmes, E. G., Fisher, R., Kleyn, M., & Ahmad, A. (2020). Current Practices for U.S. Newborn Screening of Pompe Disease and MPSI. International Journal of Neonatal Screening, 6(3), 72. https://doi.org/10.3390/ijns6030072