
Advances in the Diagnosis and Treatment of Krabbe Disease

Abstract
1. Introduction
2. GALC Purification and Cloning of the Gene
3. Early Diagnostic Studies and Mutation Analysis
4. Relationship between Measured GALC Activity and Phenotype
5. Effect of Mutations Identified and Phenotype
6. Management of Individuals Confirmed to Be at Risk for Developing Krabbe Disease
7. Treatment of the Animal Models of Krabbe Disease
Author Contributions
Funding
Conflicts of Interest
References
- Wenger, D.A.; Escolar, M.L.; Luzi, P.; Rafi, M.A. Krabbe disease (globoid cell leukodystrophy). In The Online Metabolism & Molecular Bases of Inherited Disease; Valle, D., Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Eds.; McGraw Hill: New York, NY, USA, 2013. [Google Scholar]
- Krabbe, K. A new familial, infantile form of diffuse brain-sclerosis. Brain 1916, 39, 74–114. [Google Scholar] [CrossRef]
- De Lange, C. Ueber die familiare infantile form der Gehirn-sklerose (Krabbe). Ann. Paediat. 1940, 154, 140–179. [Google Scholar]
- Collier, J.; Greenfield, J.G. The encephalitis periaxialis of schilder. A clinical and pathological study, with an account of two cases, one of which was diagnosed during life. Brain 1924, 47, 489–519. [Google Scholar] [CrossRef]
- Blackwood, W.; Cumings, J.N. A histochemical and chemical study of three cases of diffuse cerebral sclerosis. J. Neurol. Neurosurg. Psychiatry 1954, 17, 33–49. [Google Scholar] [CrossRef]
- Malone, M. Deficiency in degradative enzyme system in globoid leucodystrophy. Tran. Am. Soc. Neurochem. 1970, 1, 56. [Google Scholar]
- Suzuki, K.; Suzuki, Y. Globoid cell leucodystrophy (Krabbe’s disease): Deficiency of galactocerebroside β-galactosidase. Proc. Natl. Acad. Sci. USA 1970, 66, 302–309. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Bell, R.M. Lysosphingolipids inhibit protein kinase C: Implications for the sphingolipidoses. Science 1987, 235, 670–674. [Google Scholar] [CrossRef]
- Sugama, S.; Kim, S.U.; Ida, H.; Eto, Y. Psychosine Cytotoxicity in Rat Neural Cell Cultures and Protection by Phorbol Ester and Dimethyl Sulfoxide. Pediatr. Res. 1990, 28, 473–476. [Google Scholar] [CrossRef]
- Tanaka, K.; Webster, H.D. Effects of Psychosine (Galactosylsphingosine) on the Survival and the Fine Structure of Cultured Schwann Cells. J. Neuropathol. Exp. Neurol. 1993, 52, 490–498. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, M.W.; Kim, S.U. Tissue Culture Model of Krabbe’s Disease: Psychosine Cytotoxicity in Rat Oligodendrocyte Culture. Dev. Neurosci. 1997, 19, 321–327. [Google Scholar] [CrossRef]
- Tohyama, J.; Matsuda, J.; Suzuki, K. Psychosine Is as Potent an Inducer of Cell Death as C6-Ceramide in Cultured Fibroblasts and in MOCH-1 Cells. Neurochem. Res. 2001, 26, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Jatana, M.; Giri, S.; Singh, A.K. Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci. Lett. 2002, 330, 183–187. [Google Scholar] [CrossRef]
- Haq, E.; Giri, S.; Singh, I.; Singh, A.K. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J. Neurochem. 2003, 86, 1428–1440. [Google Scholar] [CrossRef]
- Zaka, P.; Wenger, D.A. Psychosine-induced apoptosis in a mouse oligodendrocyte progenitor cell line is mediated by capase activation. Neurosci. Lett. 2004, 358, 205–209. [Google Scholar] [CrossRef]
- Svennerholm, L.; Vanier, M.T.; Mansson, J.E. Krabbe disease: A galactosylsphingosine (psychosine) lipidosis. J. Lipid Res. 1980, 21, 53–58. [Google Scholar] [CrossRef]
- Herbst, Z.; Turgeon, C.T.; Biski, C.; Khaledi, H.; Shoemaker, N.B.; DeArmond, P.D.; Smith, S.; Orsini, J.; Matern, D.; Gelb, M.H. Achieving Congruence among Reference Laboratories for Absolute Abundance Measurement of Analytes for Rare Diseases: Psychosine for Diagnosis and Prognosis of Krabbe Disease. Int. J. Neonatal Screen. 2020, 6, 29. [Google Scholar] [CrossRef]
- Basheeruddin, K.; Shao, R.; Balster, F.; Gardley, P.; Ashbaugh, L. Newborn Screening for Krabbe Disease—Illinois Experience: Role of Psychosine in Diagnosis of the Disease. Int. J. Neonatal Screen. 2021, 7, 24. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Wenger, D.A. Galactocerebrosidase from human urine: Purification and partial characterization. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1993, 1170, 53–61. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Rafi, M.A.; De Gala, G.; Wenger, D.A. Cloning and expression cDNA encoding human galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum. Mol. Genet. 1993, 2, 1841–1846. [Google Scholar] [CrossRef]
- Luzi, P.; Rafi, M.A.; Wenger, D.A. Structure and organization of the human galactocerebrosidase (GALC) gene. Genomics 1995, 26, 407–409. [Google Scholar] [CrossRef]
- Victoria, T.; Rafi, M.A.; Wenger, D.A. Cloning of the Canine GALC cDNA and Identification of the Mutation Causing Globoid Cell Leukodystrophy in West Highland White and Cairn Terriers. Genomics 1996, 33, 457–462. [Google Scholar] [CrossRef]
- Sakai, N.; Inui, K.; Tatsumi, N.; Fukushima, H.; Nishigaki, T.; Taniike, M.; Nishimoto, J.; Tsukamoto, H.; Yanagihara, I.; Ozono, K.; et al. Molecular Cloning and Expression of cDNA for Murine Galactocerebrosidase and Mutation Analysis of the Twitcher Mouse, a Model of Krabbe’s Disease. J. Neurochem. 1996, 66, 1118–1124. [Google Scholar] [CrossRef]
- Luzi, P.; Rafi, M.A.; Victoria, T.; Baskin, G.B.; Wenger, D.A. Characterization of the Rhesus Monkey Galactocerebrosidase (GALC) cDNA and Gene and Identification of the Mutation Causing Globoid Cell Leukodystrophy (Krabbe Disease) in This Primate. Genomics 1997, 42, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nickelson, J.; Coulson, A.R. Sequencing with chain termination. Proc. Natl. Acad. Sci. USA 1997, 74, 5463–5467. [Google Scholar] [CrossRef]
- Rafi, M.A.; Luzi, P.; Chen, Y.Q.; Wenger, D.A. A large deletion together with a point mutation in the GALC gene is a common mutant allele in patients with infantile Krabbe disease. Hum. Mol. Genet. 1995, 4, 1285–1289. [Google Scholar] [CrossRef]
- Luzi, P.; Rafi, M.A.; Wenger, D.A. Characterization of the large deletion in the GALC gene found in patients with Krabbe disease. Hum. Mol. Genet. 1995, 4, 2335–2338. [Google Scholar] [CrossRef]
- Liao, H.-C.; Spacil, Z.; Ghomashchi, F.; Escolar, M.L.; Kurtzberg, J.; Orsini, J.J.; Turecek, F.; Scott, C.R.; Gelb, M.H. Lymphocyte Galactocerebrosidase Activity by LC-MS/MS for Post–Newborn Screening Evaluation of Krabbe Disease. Clin. Chem. 2017, 63, 1363–1369. [Google Scholar] [CrossRef]
- Wenger, D.A.; Riccardi, V.M. Possible misdiagnosis of Krabbe disease. J. Pediatr. 1976, 88, 76–79. [Google Scholar] [CrossRef]
- Wenger, D.A.; Luzi, P.; Rafi, M.A. Krabbe disease: Are certain mutations disease-causing only when specific polymorphisms are present or when inherited in trans with specific second mutations? Mol. Genet. Metab. 2014, 111, 307–308. [Google Scholar] [CrossRef]
- Saavedra-Matiz, C.A.; Luzi, P.; Nichols, M.; Orsini, J.J.; Caggana, M.; Wenger, D.A. Expression of individual mutations and haplotypes in the GALC gene identified by The Newborn Screening Program in New York State and in confirmed cases of Krabbe disease. J. Neurosci. Res. 2016, 94, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Duffner, P.K.; Caggana, M.; Orsini, J.J.; Wenger, D.A.; Patterson, M.; Crosley, C.J.; Kurtzberg, J.; Arnold, G.L.; Escolar, M.L.; Adams, D.; et al. Newborn Screening for Krabbe Disease: The New York State Model. Pediatr. Neurol. 2009, 40, 245–252. [Google Scholar] [CrossRef]
- Spratley, S.J.; Hill, C.H.; Viuff, A.H.; Edgar, J.R.; Skjødt, K.; Deane, J.E. Molecular Mechanisms of Disease Pathogenesis Differ in Krabbe Disease Variants. Traffic 2016, 17, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.L.; Herrera, D.G.; Rn, D.C.; DeGasoperi, R.; Sosa, M.A.G.; Kolodny, E.H.; Trifiletti, R. Adult-onset Krabbe’s disease in siblings with novel mutations in the galactocerebrosidase gene. Ann. Neurol. 1997, 41, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, K.; Gelb, M.H.; Grantham, A.; Spencer, N.; Webb, C.; West, T. Family attitudes regarding newborn screening for Krabbe disesase: Results from a survey of leukodystrophy registries. Int. J. Neonatal. Screen. 2020, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Escolar, M.L.; Poe, M.D.; Provenzale, J.M.; Richards, K.C.; Allison, J.; Wood, S.; Wenger, D.A.; Pietryga, D.; Wall, D.; Champagne, M.; et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N. Engl. J. Med. 2005, 352, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Krivit, W.; Shapiro, E.G.; Peters, C.; Wagner, J.E.; Cornu, G.; Kurtzberg, J.; Wenger, D.A.; Kolodny, E.H.; Vanier, M.T.; Loes, D.J.; et al. Hematopoietic Stem-Cell Transplantation in Globoid-Cell Leukodystrophy. N. Engl. J. Med. 1998, 338, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.D.; Poe, M.D.; DeRenzo, A.; Haldal, S.; Escolar, M.L. Developmental outcomes of cord blood transplantation for Krabbe disease: A 15-year study. Neurology 2017, 89, 1365–1372. [Google Scholar] [CrossRef]
- Yeager, A.; Brennan, S.; Tiffany, C.; Moser, H.; Santos, G. Prolonged survival and remyelination after hematopoietic cell transplantation in the twitcher mouse. Science 1984, 225, 1052–1054. [Google Scholar] [CrossRef]
- LeVine, S.M.; Pedchenko, T.V.; Bronshteyn, I.G.; Pinson, D.M. L-cycloserine slows the clinical and pathological course in mice with globoid cell leukodystrophy (twitcher mice). J. Neurosci. Res. 2000, 60, 231–236. [Google Scholar] [CrossRef]
- Wu, Y.-P.; McMahon, E.J.; Matsuda, J.; Suzuki, K.; Matsushima, G.K.; Suzuki, K. Expression of immune-related molecules is downregulated in twitcher mice following bone marrow transplantation. J. Neuropathol. Exp. Neurol. 2001, 60, 1062–1074. [Google Scholar] [CrossRef]
- Biswas, S.; Le Vine, S.M. Substrate-Reduction Therapy Enhances the Benefits of Bone Marrow Transplantation in Young Mice with Globoid Cell Leukodystrophy. Pediatr. Res. 2002, 51, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Rafi, M.A.; Rao, H.Z.; Passini, M.A.; Curtis, M.; Vanier, M.T.; Zaka, M.; Luzi, P.; Wolfe, J.H.; Wenger, D.A. AAV-Mediated expression of galactocerebrosidase in brain results in attenuated symptoms and extended life span in murine models of globoid cell leukodystrophy. Mol. Ther. 2005, 11, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Tsoi, Y.K.; Troendle, F.J.; DeLucia, M.W.; Ahmed, Z.; Dicky, C.A.; Dickson, D.W.; Eckman, C.B. Single-dose intracerebroventricular administration of galactocerebrosidase improves survival in a mouse model of globoid cell leukodystrophy. FASEB J. 2007, 21, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Donsante, A.; Macauley, S.; Levy, B.; Vogler, C.; Sands, M.S. Central nervous system-directed AAV2/5-mediated gene therapy synergizes with bone marrow transplantation in the murine model of globoid-cell leukodystrophy. Mol. Ther. 2007, 15, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Strazza, M.; Luddi, A.; Carbone, M.; Rafi, M.A.; Costantino-Ceccarini, E.; Wenger, D.A. Significant correction of pathology in brains of twitcher mice following injection of genetically modified mouse neural progenitor cells. Mol. Genet. Metab. 2009, 97, 27–34. [Google Scholar] [CrossRef]
- Reddy, A.S.; Kim, J.H.; Hawkins-Salsbury, J.A.; Macauley, S.L.; Tracy, E.T.; Vogler, C.A.; Han, X.; Song, S.K.; Wozniak, D.F.; Fowler, S.C.; et al. Bone marrow transplantation augments the effect of brain- and spinal cord-directed adeno-associated virus 2/5 gene therapy by altering inflammation in the murine model of globoid-cell leukodystrophy. J. Neurosci. 2011, 31, 9945–9957. [Google Scholar] [CrossRef]
- Qin, E.Y.; Hawkins-Salsbury, J.A.; Jiang, X.; Reddy, A.S.; Farber, N.; Ory, D.S.; Sands, M.S. Bone marrow transplantation increases efficacy of central nervous system-directed enzyme replacement therapy in the murine model of globoid cell leukodystrophy. Mol. Genet. Metab. 2012, 107, 186–196. [Google Scholar] [CrossRef]
- Rafi, M.A.; Rao, H.Z.; Luzi, P.; Curtis, M.T.; A Wenger, D. Extended Normal Life After AAVrh10-mediated Gene Therapy in the Mouse Model of Krabbe Disease. Mol. Ther. 2012, 20, 2031–2042. [Google Scholar] [CrossRef]
- Berardi, A.S.; Pannuzzo, G.; Graziano, A.; Costantino-Ceccarini, E.; Piomboni, P.; Luddi, A. Pharmacological chaperones increase residual beta-galactocerebrosidase activity in fibroblasts from Krabbe patients. Mol. Genet. Metab. 2014, 112, 294–301. [Google Scholar] [CrossRef]
- Rafi, M.A.; Rao, H.Z.; Luzi, P.; Luddi, A.; Curtis, M.T.; Wenger, D.A. Intravenous injection of AAVrh10-GALC after the neonatal period in twitcher mice results in significant expression in the central and peripheral nervous systems and improvement of clinical features. Mol. Genet. Metab. 2015, 114, 459–466. [Google Scholar] [CrossRef]
- Rafi, M.A.; Rao, H.Z.; Luzi, P.; A Wenger, D. Long-term Improvements in Lifespan and Pathology in CNS and PNS After BMT Plus One Intravenous Injection of AAVrh10-GALC in Twitcher Mice. Mol. Ther. 2015, 23, 1681–1690. [Google Scholar] [CrossRef]
- Hawkins-Salsbury, J.A.; Shea, L.; Jiang, X.; Hunter, D.A.; Guzman, A.M.; Reddy, A.S.; Qin, E.Y.; Li, Y.; Gray, S.J.; Ory, D.S.; et al. Mechanism-Based Combination Treatment Dramatically Increases Therapeutic Efficacy in Murine Globoid Cell Leukodystrophy. J. Neurosci. 2015, 35, 6495–6505. [Google Scholar] [CrossRef] [PubMed]
- Ricca, A.; Rufo, N.; Ungari, S.; Morena, F.; Martino, S.; Kulik, W.; Alberizzi, V.; Bolino, A.; Bianchi, F.; Del Carro, U.; et al. Combined gene/cell therapies provide long-term and pervasive rescue of multiple pathological symptoms in a murine model of globoid cell leukodystrophy. Hum. Mol. Genet. 2015, 24, 3372–3389. [Google Scholar] [CrossRef] [PubMed]
- Karumuthil-Melethil, S.; Marshall, M.S.; Heindel, C.; Jakubauskas, B.; Bongarzone, E.R.; Gray, S.J. Intrathecal administration of AAV/GALC vectors in 10-11-day-old twitcher mice improves survival and is enhanced by bone marrow transplant. J. Neurosci. Res. 2016, 94, 1138–1151. [Google Scholar] [CrossRef]
- Marshall, M.S.; Issa, Y.; Jakubauskas, B.; Stoskute, M.; Elackattu, V.; Marshall, J.N.; Bogue, W.; Nguyen, D.; Hauck, Z.; Rue, E.; et al. Long-Term Improvement of Neurological Signs and Metabolic Dysfunction in a Mouse Model of Krabbe’s Disease after Global Gene Therapy. Mol. Ther. 2018, 26, 874–889. [Google Scholar] [CrossRef]
- Rafi, M.A.; Luzi, P.; Wenger, D.A. Conditions for combining gene therapy with bone marrow transplantation in murine Krabbe disease. BioImpacts 2020, 10, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Luzi, P.; Abraham, R.M.; Rafi, M.A.; Curtis, M.; Hooper, D.C.; Wenger, D.A. Effects of treatments on inflammatory and apoptotic markers in the CNS of mice with globoid cell leukodystrophy. Brain Res. 2009, 1300, 146–158. [Google Scholar] [CrossRef][Green Version]
- Rafi, M.A.; Luzi, P.; A Wenger, D. Can early treatment of twitcher mice with high dose AAVrh10-GALC eliminate the need for BMT? BioImpacts 2021, 11, 135–146. [Google Scholar] [CrossRef]
- Bradbury, A.M.; Rafi, M.A.; Bagel, J.H.; Brisson, B.K.; Marshall, M.S.; Salvador, J.P.; Jiang, X.; Swain, G.P.; Prociuk, M.L.; Odonnell, P.A.; et al. AAVrh10 Gene Therapy Ameliorates Central and Peripheral Nervous System Disease in Canine Globoid Cell Leukodystrophy (Krabbe Disease). Hum. Gene Ther. 2018, 29, 785–801. [Google Scholar] [CrossRef]


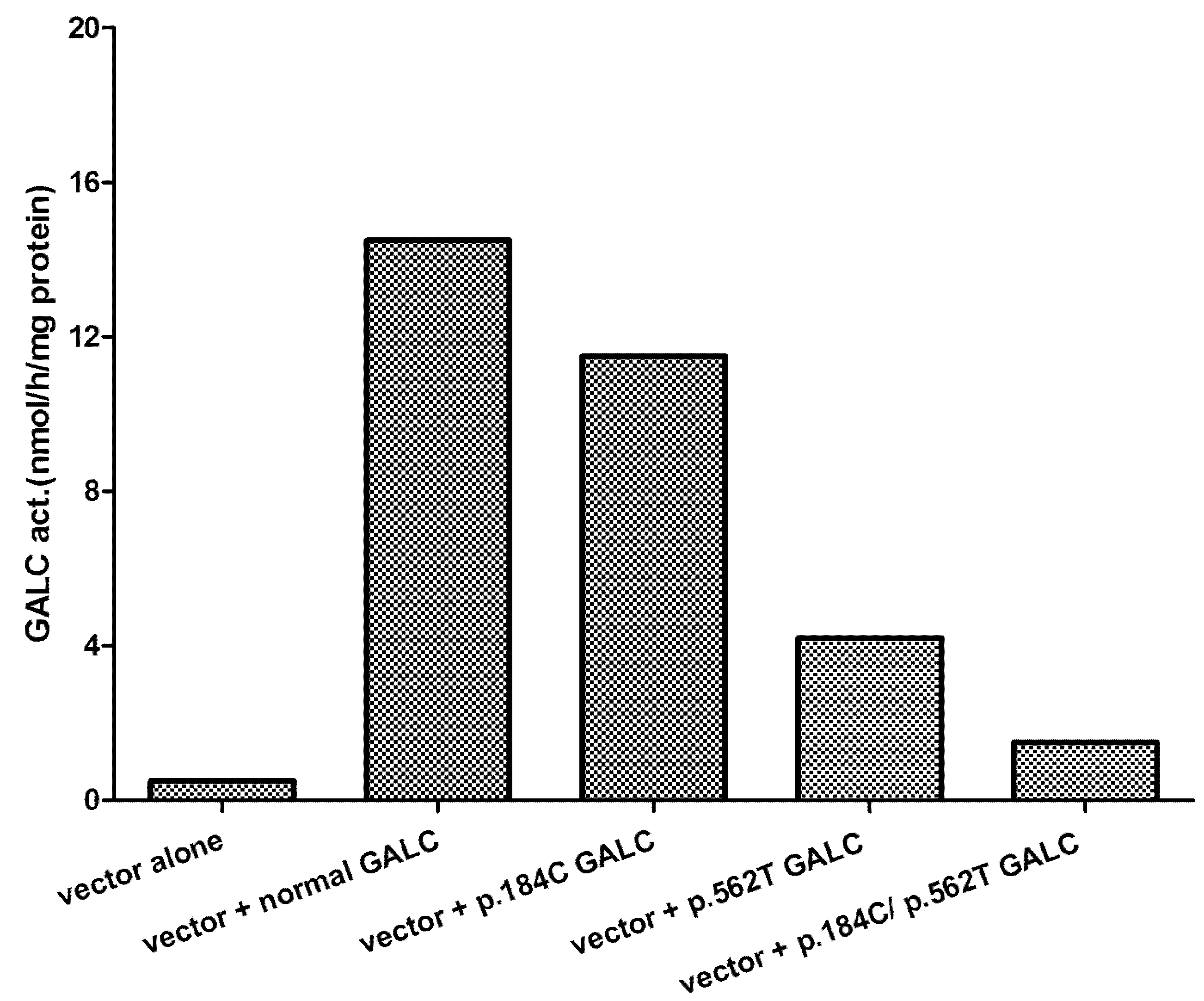{kind=link}
| Location | Nucleotide Change in cDNA | Codon Change | Effect | Comments b |
|---|---|---|---|---|
| Ex 1 | c.121G>A (c.169G>A) a | GGC>AGC | p.G41S (p.G57S) | Mild, Southern Italy |
| Ex 4 | c.284G>A (c.332G>A) | GGC>GAC | p.G95D (p.G111D) | Severe |
| Ex 4 | c.286A>G (c.334A>G) | ACT>GCT | p.T96A (p.T112A) | Mild c |
| Ex 7 | c.635 del + ins (c.683 del + ins) | del 12, ins3 | del 5 aa + ins 2 aa | Severe, Japanese, Korean |
| Ex 8 | c.809G>A (c.857G>A) | GGC>GAC | p.G270D (p.G286D) | Mild |
| Ex 8 | c.860C>T (c.908C>T) | TCC>TTC | p.S287F (p.S303F) | Severe |
| Ex 9 | c.908A>G (c.956A>G) | TAT>TGT | p.Y303C (p.Y319C) | Mild d |
| In10-end | 30 kb deletion | 30 kb del | Short mRNA | Severe |
| Ex 11 | c.1138C>T (c.1186C>T) | CGG>TGG | p.R380W (p.R396W) | Severe |
| Ex 13 | c.1424 delA (c.1472 delA) | TAAGG>TAGG | FS, PS | Severe |
| Ex 14 | c.1538 C>T (c.1586C>T) | ACG>ATG | p.T513M (p.T529M) | Severe |
| Ex 15 | c.1652A>C (c.1700A>C) | TAC>TCC | p.Y551S (p.Y567S) | Severe |
| Ex 16 | c.1853T>C) (c.1901T>C) | TTA>TCA | p.L618S (p.L634S) | Mild (Asian) |
| Location | Nucleotide Change in cDNA | Codon Change | Effect | Comments b |
|---|---|---|---|---|
| Ex 4 | c.502C>T (c.550C>T) a | CGT>TGT | p.R168C (p.R184C) | The 30 kb deletion always has this polymorphism |
| Ex 6 | c.694G>A (c.742G>A) | GAT>AAT | p.D232N (p.D248N) | |
| Ex 8 | c.865A>G (c.913A>G) | ATC>GTC | p.I289V (p.I305V) | Japanese |
| Ex 14 | c.1637T>C (c.1685T>C) | ATA>ACA | p.I546T (p.I562T) | Very common |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wenger, D.A.; Luzi, P.; Rafi, M.A. Advances in the Diagnosis and Treatment of Krabbe Disease. Int. J. Neonatal Screen. 2021, 7, 57. https://doi.org/10.3390/ijns7030057
Wenger DA, Luzi P, Rafi MA. Advances in the Diagnosis and Treatment of Krabbe Disease. International Journal of Neonatal Screening. 2021; 7(3):57. https://doi.org/10.3390/ijns7030057
Chicago/Turabian StyleWenger, David A, Paola Luzi, and Mohammad A. Rafi. 2021. "Advances in the Diagnosis and Treatment of Krabbe Disease" International Journal of Neonatal Screening 7, no. 3: 57. https://doi.org/10.3390/ijns7030057
APA StyleWenger, D. A., Luzi, P., & Rafi, M. A. (2021). Advances in the Diagnosis and Treatment of Krabbe Disease. International Journal of Neonatal Screening, 7(3), 57. https://doi.org/10.3390/ijns7030057