
DBS Screening for Glycogen Storage Disease Type 1a: Detection of c.648G>T Mutation in G6PC by Combination of Modified Competitive Oligonucleotide Priming-PCR and Melting Curve Analysis

 ,
,  , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patients and Controls
2.1.1. Informed Consents and Ethics Committee Approval
2.1.2. Patient Clinical Information
2.2. Detection of G6PC and CFTR
2.2.1. Preparation of DBS Samples from Controls and Patients
2.2.2. Construction of Carrier Status Model
2.2.3. Outline of PCR Detection System for c.648G>T in G6PC
- First-round PCR amplification of G6PC intron 4–exon 5 and CFTR exon 4–intron 4 using the outer primer sets was done by conventional PCR. A small, punched circle from the DBS was used directly for PCR, without any DNA extraction or purification procedures.
- Second-round PCR amplification of G6PC intron 4–exon 5 and CFTR exon 4 using the inner primer sets was done by real-time mCOP-PCR. The two alleles, c.648G (wild type) and c.648T (mutant), were specifically amplified in this step.
- Melting curve analysis of the amplification products was immediately started after the second-round PCR amplification ended. The G6PC peaks, c.648G and c.648T, were clearly separated from the CFTR peaks.
2.2.4. First-Round PCR: Multiplex Amplification of G6PC and CFTR Outer Fragments
2.2.5. Second-Round PCR: Multiplex Amplification of G6PC and CFTR Inner Fragments
2.2.6. Melting Curve Analysis
2.3. Sequencing Analysis
2.4. Statistical Analysis
3. Results
3.1. First-Round PCR Followed by Gel Electrophoresis
3.2. Second-Round PCR Followed by Gel Electrophoresis
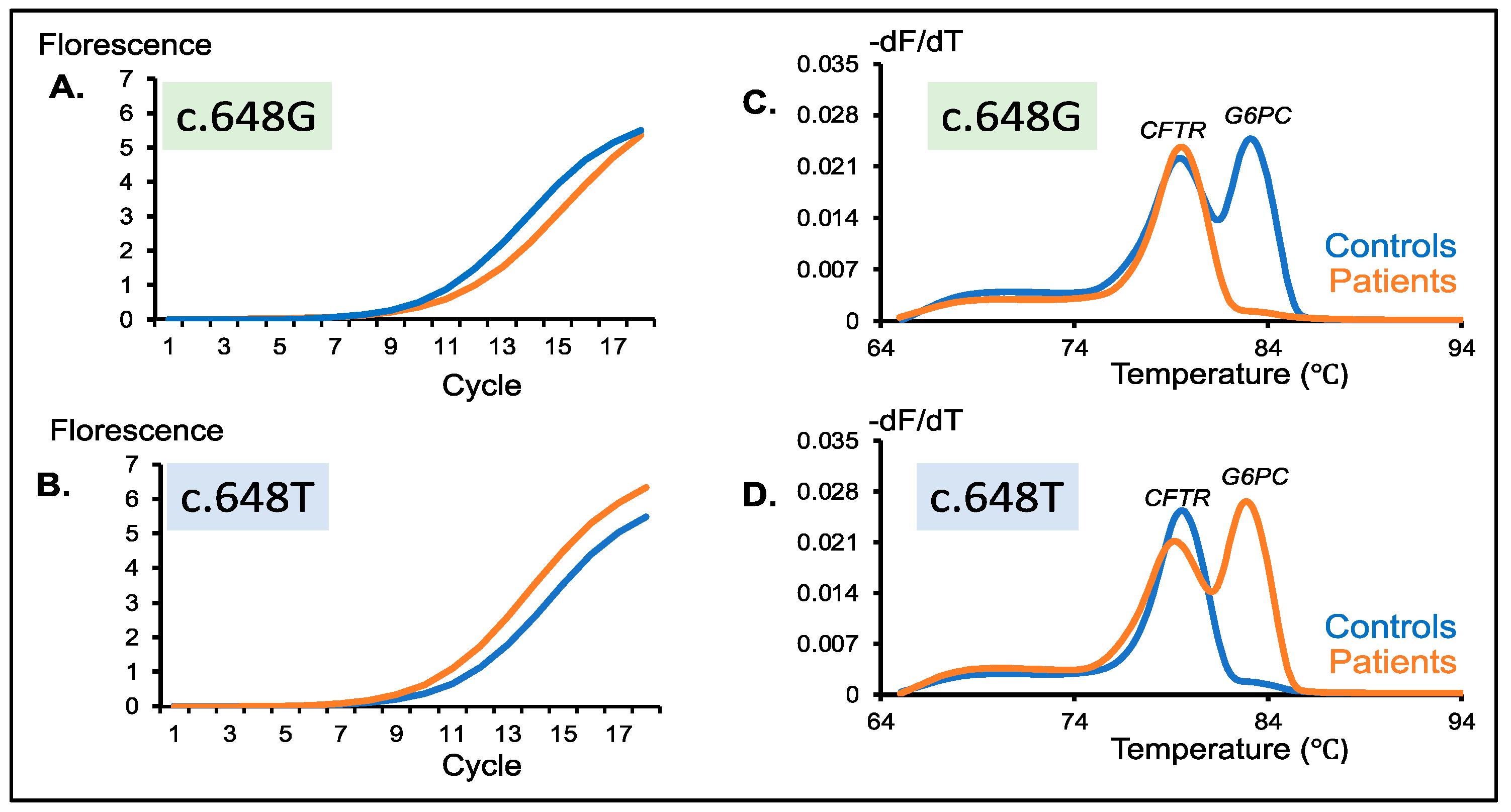
3.3. Amplification Curve Analysis of the Second-Round PCR Products
3.4. Melting Curve Analysis of the Second-Round PCR Products
3.5. GSD1a Carrier Model Screening for GSDIa
3.6. Peak Height Ratio of G6PC vs. CFTR
4. Discussion
4.1. Necessity of GSD1a Screening in the Context of Gene Replacement Therapy
4.2. Nested mCOP-PCR as a New Screening System to Detect c.648G>T in G6PC
4.3. Screening Strategy for GSDIa in the Real World
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cori, G.T.; Cori, C.F. Glucose-6-Phosphatase of the Liver in Glycogen Storage Disease. J. Biol. Chem. 1952, 199, 661–667. [Google Scholar] [CrossRef]
- Beyzaei, Z.; Geramizadeh, B. Molecular Diagnosis of Glycogen Storage Disease Type I: A Review. EXCLI J. 2019, 18, 30–46. [Google Scholar] [PubMed]
- Ekstein, J.; Rubin, B.Y.; Anderson, S.L.; Weinstein, D.A.; Bach, G.; Abeliovich, D.; Webb, M.; Risch, N. Mutation Frequencies for Glycogen Storage Disease Ia in the Ashkenazi Jewish Population. Am. J. Med. Genet. 2004, 129A, 162–164. [Google Scholar] [CrossRef]
- Lei, K.J.; Pan, C.J.; Shelly, L.L.; Liu, J.L.; Chou, J.Y. Identification of Mutations in the Gene for Glucose-6-Phosphatase, the Enzyme Deficient in Glycogen Storage Disease Type 1a. J. Clin. Investig. 1994, 93, 1994–1999. [Google Scholar] [CrossRef]
- Human Gene Mutation Database. Available online: http://www.Hgmd.Cf.Ac.Uk/Ac/Index.Php (accessed on 9 October 2020).
- Lei, K.J.; Chen, Y.T.; Chen, H.; Wong, L.J.; Liu, J.L.; McConkie-Rosell, A.; Van Hove, J.L.; Ou, H.C.; Yeh, N.J.; Pan, L.Y. Genetic Basis of Glycogen Storage Disease Type 1a: Prevalent Mutations at the Glucose-6-Phosphatase Locus. Am. J. Hum. Genet. 1995, 57, 766–771. [Google Scholar]
- Akanuma, J.; Nishigaki, T.; Fujii, K.; Matsubara, Y.; Inui, K.; Takahashi, K.; Kure, S.; Suzuki, Y.; Ohura, T.; Miyabayashi, S.; et al. Glycogen Storage Disease Type Ia: Molecular Diagnosis of 51 Japanese Patients and Characterization of Splicing Mutations by Analysis of Ectopically Transcribed MRNA from Lymphoblastoid Cells. Am. J. Med. Genet. 2000, 91, 107–112. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Choi, J.-H.; Lee, B.-H.; Kim, G.-H.; Kim, K.-M.; Yoo, H.-W. Predominance of the c.648G>T G6PC Gene Mutation and Late Complications in Korean Patients with Glycogen Storage Disease Type Ia. Orphanet. J. Rare Dis. 2020, 15, 45. [Google Scholar] [CrossRef]
- Kajihara, S.; Matsuhashi, S.; Yamamoto, K.; Kido, K.; Tsuji, K.; Tanae, A.; Fujiyama, S.; Itoh, T.; Tanigawa, K.; Uchida, M. Exon Redefinition by a Point Mutation within Exon 5 of the Glucose-6-Phosphatase Gene Is the Major Cause of Glycogen Storage Disease Type 1a in Japan. Am. J. Hum. Genet. 1995, 57, 549–555. [Google Scholar] [CrossRef]
- Lam, C.-W.; But, W.-M.; Shek, C.-C.; Tong, S.-F.; Chan, Y.-S.; Choy, K.-W.; Tse, W.-Y.; Pang, C.-P.; Hjelm, N.M. Glucose-6-Phosphatase Gene (727G→T) Splicing Mutation Is Prevalent in Hong Kong Chinese Patients with Glycogen Storage Disease Type La. Clin. Genet. 2008, 53, 184–190. [Google Scholar] [CrossRef]
- Yang Chou, J. The Molecular Basis of Type 1 Glycogen Storage Diseases. Curr. Mol. Med. 2001, 1, 25–44. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and Management of Glycogen Storage Disease Type I: A Practice Guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014, 16, e1. [Google Scholar] [CrossRef]
- Melis, D.; Pivonello, R.; Cozzolino, M.; Della Casa, R.; Balivo, F.; Del Puente, A.; Dionisi-Vici, C.; Cotugno, G.; Zuppaldi, C.; Rigoldi, M.; et al. Impaired Bone Metabolism in Glycogen Storage Disease Type 1 Is Associated with Poor Metabolic Control in Type 1a and with Granulocyte Colony-Stimulating Factor Therapy in Type 1b. Horm. Res. Paediatr. 2014, 81, 55–62. [Google Scholar] [CrossRef]
- Romano, A.; Russo, D.; Contaldo, M.; Lauritano, D.; della Vella, F.; Serpico, R.; Lucchese, A.; Stasio, D.D. Oral Manifestations in Patients with Glycogen Storage Disease: A Systematic Review of the Literature. Appl. Sci. 2020, 10, 6720. [Google Scholar] [CrossRef]
- Kudo, M. Hepatocellular Adenoma in Type Ia Glycogen Storage Disease. J. Gastroenterol. 2001, 36, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Sharif, K.; Brown, R.M.; Morland, B. Hepatocellular Carcinoma in Children. Clin. Liver Dis. 2015, 19, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Aoun, B.; Sanjad, S.; Degheili, J.A.; Barhoumi, A.; Bassyouni, A.; Karam, P.E. Kidney and Metabolic Phenotypes in Glycogen Storage Disease Type-I Patients. Front. Pediatrics 2020, 8, 591. [Google Scholar] [CrossRef]
- Chou, J.Y.; Kim, G.-Y.; Cho, J.-H. Recent Development and Gene Therapy for Glycogen Storage Disease Type Ia. Liver Res. 2017, 1, 174–180. [Google Scholar] [CrossRef]
- Chou, J.; Zingone, A.; Pan, C.-J. Adenovirus-Mediated Gene Therapy in a Mouse Model of Glycogen Storage Disease Type 1a. Eur. J. Pediatrics 2002, 161, S56–S61. [Google Scholar] [CrossRef]
- Kim, G.-Y.; Lee, Y.M.; Kwon, J.H.; Cho, J.-H.; Pan, C.-J.; Starost, M.F.; Mansfield, B.C.; Chou, J.Y. Glycogen Storage Disease Type Ia Mice with Less than 2% of Normal Hepatic Glucose-6-Phosphatase-α Activity Restored Are at Risk of Developing Hepatic Tumors. Mol. Genet. Metab. 2017, 120, 229–234. [Google Scholar] [CrossRef]
- Roseman, D.S.; Khan, T.; Rajas, F.; Jun, L.S.; Asrani, K.H.; Isaacs, C.; Farelli, J.D.; Subramanian, R.R. G6PC MRNA Therapy Positively Regulates Fasting Blood Glucose and Decreases Liver Abnormalities in a Mouse Model of Glycogen Storage Disease 1a. Mol. Ther. 2018, 26, 814–821. [Google Scholar] [CrossRef]
- Hinderer, C.; Bell, P.; Louboutin, J.-P.; Zhu, Y.; Yu, H.; Lin, G.; Choa, R.; Gurda, B.L.; Bagel, J.; O’Donnell, P.; et al. Neonatal Systemic AAV Induces Tolerance to CNS Gene Therapy in MPS I Dogs and Nonhuman Primates. Mol. Ther. 2015, 23, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.M.; Aghili, A.; Li, S.; Ongoiba, A.; Kayentao, K.; Doumbo, S.; Traore, B.; Crompton, P.D. A Nested Real-Time PCR Assay for the Quantification of Plasmodium Falciparum DNA Extracted from Dried Blood Spots. Malar. J. 2014, 13, 393. [Google Scholar] [CrossRef] [PubMed]
- Wijaya, Y.O.S.; Purevsuren, J.; Harahap, N.I.F.; Niba, E.T.E.; Bouike, Y.; Nurputra, D.K.; Rochmah, M.A.; Thursina, C.; Hapsara, S.; Yamaguchi, S.; et al. Assessment of Spinal Muscular Atrophy Carrier Status by Determining SMN1 Copy Number Using Dried Blood Spots. Int. J. Neonatal Screen. 2020, 6, 43. [Google Scholar] [CrossRef]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Samaranch, L.; Sebastian, W.S.; Kells, A.P.; Salegio, E.A.; Heller, G.; Bringas, J.R.; Pivirotto, P.; DeArmond, S.; Forsayeth, J.; Bankiewicz, K.S. AAV9-Mediated Expression of a Non-Self Protein in Nonhuman Primate Central Nervous System Triggers Widespread Neuroinflammation Driven by Antigen-Presenting Cell Transduction. Mol. Ther. 2014, 22, 329–337. [Google Scholar] [CrossRef]
- Monahan, P.E.; Négrier, C.; Tarantino, M.; Valentino, L.A.; Mingozzi, F. Emerging Immunogenicity and Genotoxicity Considerations of Adeno-Associated Virus Vector Gene Therapy for Hemophilia. JCM 2021, 10, 2471. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Bell, P.; Louboutin, J.-P.; Katz, N.; Zhu, Y.; Lin, G.; Choa, R.; Bagel, J.; O’Donnell, P.; Fitzgerald, C.A.; et al. Neonatal Tolerance Induction Enables Accurate Evaluation of Gene Therapy for MPS I in a Canine Model. Mol. Genet. Metab. 2016, 119, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Lotfinia, M.; Abdollahpour-Alitappeh, M.; Hatami, B.; Zali, M.R.; Karimipoor, M. Adeno-Associated Virus as a Gene Therapy Vector: Strategies to Neutralize the Neutralizing Antibodies. Clin. Exp. Med. 2019, 19, 289–298. [Google Scholar] [CrossRef]
- Li, C.; Narkbunnam, N.; Samulski, R.J.; Asokan, A.; Hu, G.; Jacobson, L.J.; Manco-Johnson, M.J.; Monahan, P.E. Neutralizing Antibodies against Adeno-Associated Virus Examined Prospectively in Pediatric Patients with Hemophilia. Gene Ther. 2012, 19, 288–294. [Google Scholar] [CrossRef]
- Mimuro, J.; Mizukami, H.; Shima, M.; Matsushita, T.; Taki, M.; Muto, S.; Higasa, S.; Sakai, M.; Ohmori, T.; Madoiwa, S.; et al. The Prevalence of Neutralizing Antibodies against Adeno-Associated Virus Capsids Is Reduced in Young Japanese Individuals: Prevalence of Antibodies Against AAV. J. Med. Virol. 2014, 86, 1990–1997. [Google Scholar] [CrossRef]
- Cao, J.; Choi, M.; Guadagnin, E.; Soty, M.; Silva, M.; Verzieux, V.; Weisser, E.; Markel, A.; Zhuo, J.; Liang, S.; et al. MRNA Therapy Restores Euglycemia and Prevents Liver Tumors in Murine Model of Glycogen Storage Disease. Nat. Commun. 2021, 12, 3090. [Google Scholar] [CrossRef]
- Ar Rochmah, M.; Harahap, N.I.F.; Niba, E.T.E.; Nakanishi, K.; Awano, H.; Morioka, I.; Iijima, K.; Saito, T.; Saito, K.; Lai, P.S.; et al. Genetic Screening of Spinal Muscular Atrophy Using a Real-Time Modified COP-PCR Technique with Dried Blood-Spot DNA. Brain Dev. 2017, 39, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Niba, E.T.E.; Wijaya, Y.O.S.; Takayama, I.; Mitsuishi, C.; Kumasaka, S.; Kondo, Y.; Takatera, A.; Hokuto, I.; Morioka, I.; et al. A Novel System for Spinal Muscular Atrophy Screening in Newborns: Japanese Pilot Study. Int. J. Neonatal Screen. 2019, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Niba, E.T.E.; Rochmah, M.A.; Harahap, N.I.F.; Awano, H.; Morioka, I.; Iijima, K.; Takeshima, Y.; Saito, T.; Saito, K.; Takeuchi, A.; et al. Spinal Muscular Atrophy: New Screening System with Real-Time MCOP-PCR and PCR-RFLP for SMN1 Deletion. Kobe J. Med. Sci. 2019, 65, E44–E48. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Ratios | Mean | SD |
|---|---|---|---|
| Control DBS (n = 50) | G-ratio (G6PC [G]/CFTR) | 1.20 | ± 0.30 |
| T-ratio (G6PC [T]/CFTR) | 0.06 | ± 0.02 | |
| Patient DBS (n = 4) | G-ratio (G6PC [G]/CFTR) | 0.08 | ± 0.02 |
| T-ratio (G6PC [T]/CFTR) | 1.43 | ± 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niba, E.T.E.; Wijaya, Y.O.S.; Awano, H.; Taniguchi, N.; Takeshima, Y.; Nishio, H.; Shinohara, M. DBS Screening for Glycogen Storage Disease Type 1a: Detection of c.648G>T Mutation in G6PC by Combination of Modified Competitive Oligonucleotide Priming-PCR and Melting Curve Analysis. Int. J. Neonatal Screen. 2021, 7, 79. https://doi.org/10.3390/ijns7040079
Niba ETE, Wijaya YOS, Awano H, Taniguchi N, Takeshima Y, Nishio H, Shinohara M. DBS Screening for Glycogen Storage Disease Type 1a: Detection of c.648G>T Mutation in G6PC by Combination of Modified Competitive Oligonucleotide Priming-PCR and Melting Curve Analysis. International Journal of Neonatal Screening. 2021; 7(4):79. https://doi.org/10.3390/ijns7040079
Chicago/Turabian StyleNiba, Emma Tabe Eko, Yogik Onky Silvana Wijaya, Hiroyuki Awano, Naoko Taniguchi, Yasuhiro Takeshima, Hisahide Nishio, and Masakazu Shinohara. 2021. "DBS Screening for Glycogen Storage Disease Type 1a: Detection of c.648G>T Mutation in G6PC by Combination of Modified Competitive Oligonucleotide Priming-PCR and Melting Curve Analysis" International Journal of Neonatal Screening 7, no. 4: 79. https://doi.org/10.3390/ijns7040079
APA StyleNiba, E. T. E., Wijaya, Y. O. S., Awano, H., Taniguchi, N., Takeshima, Y., Nishio, H., & Shinohara, M. (2021). DBS Screening for Glycogen Storage Disease Type 1a: Detection of c.648G>T Mutation in G6PC by Combination of Modified Competitive Oligonucleotide Priming-PCR and Melting Curve Analysis. International Journal of Neonatal Screening, 7(4), 79. https://doi.org/10.3390/ijns7040079