
Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing


Abstract
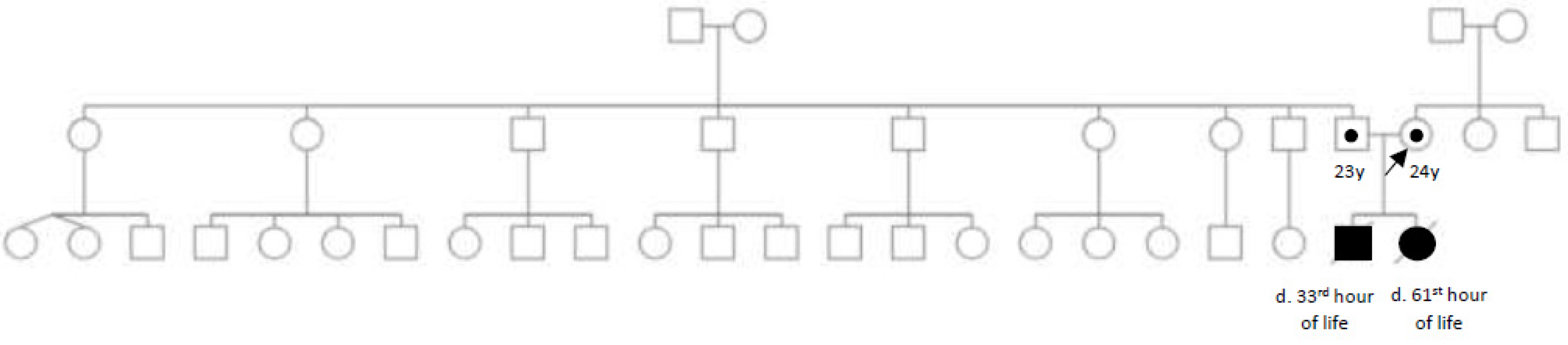
:1. Introduction
2. Case Report
3. Investigations
3.1. Biochemical Tests
3.2. Genetic Tests
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iacobazzi, V.; Invernizzi, F.; Baratta, S.; Pons, R.; Chung, W.; Garavaglia, B.; Dionisi-Vici, C.; Ribes, A.; Parini, R.; Dolores Huertas, M.; et al. Molecular and functional analysis of SLC25A20 mutations causing carnitine-acylcarnitine translocase deficiency. Hum. Mutat. 2004, 24, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Gozalbo, M.; Bakker, J.; Waterham, H.; Wanders, R. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol. Asp. Med. 2004, 25, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.-M.; Hu, H.; Ahmed, A.; Feng, B.-B.; Liu, J.; Jia, Z.-J.; Wang, H. Carnitine-acylcarnitine translocase deficiency with c.199-10 t>g and novel c.1a>g mutation. Medicine 2017, 96, e8549. [Google Scholar] [CrossRef] [PubMed]
- Vitoria, I.; Martín-Hernández, E.; Peña-Quintana, L.; Bueno, M.; Quijada-Fraile, P.; Dalmau, J.; Molina-Marrero, S.; Pérez, B.; Merinero, B. Carnitineacylcarnitine translocase deficiency: Experience with four cases in Spain and review of the literature. JIMD Rep. 2015, 20, 11–20. [Google Scholar] [PubMed] [Green Version]
- Hsu, B.Y.; Iacobazzi, V.; Wang, Z.; Harvie, H.; Chalmers, R.; Saudubray, J.; Palmieri, F.; Ganguly, A.; Stanley, C. Aberrant mRNA splicing associated with coding region mutations in children with carnitine-acylcarnitine translocase deficiency. Mol. Genet. Metab. 2001, 74, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Hoffmann, G.F.; Matern, D. Newborn screening for disorders of fatty-acid oxidation: Experience and recommendations from an expert meeting. J. Inherit. Metab. Dis. 2010, 33, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.; Tang, N.L.; Li, C.K.; Law, L.K.; To, K.F.; Yau, P.; Fung, S.L.M.; Chong, J.S.C.; Tsung, L.; Chiang, G.; et al. Inherited metabolic diseases in the Southern Chinese population: Spectrum of diseases and estimated incidence from recurrent mutations. Pathology 2014, 46, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Cai, Y.; Li, S.; Xiong, H.; Liu, M.; Ma, F.; Xiao, X.; Hao, H. Late-Onset Carnitine–Acylcarnitine Translocase Deficiency with SLC25A20 c.199-10T>G Variation: Case Report and Pathologic Analysis of Liver Biopsy. Front. Pediatr. 2020, 8, 585646. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.S.; Lam, C.W.; Lai, C.K.; Yuen, Y.P.; Chan, K.Y.; Shek, C.C.; Chan, A.Y.W.; Chow, C.B. Carnitineacylcarnitine translocase deficiency in three neonates presenting with rapid deterioration and cardiac arrest. Hong Kong Med. J. 2007, 13, 66–68. [Google Scholar] [PubMed]
- Hoffmann, G.I.; Nyhan, W.L.; Zschacke, J.; Kahler, S.G.; Mayatepek, E. (Eds.) Inherited Metabolic Diseases, 1st ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2002; p. 82. [Google Scholar]
- Costa, C.; Costa, J.M.; Slama, A.; Boutron, A.; Vequaud, C.; Legrand, A.; Brivet, M. Mutational spectrum and DNA-based prenatal diagnosis in carnitine-acyl carnitine translocate deficiency. Mol. Genet. Metab. 2003, 78, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Vatanavicharn, N.; Yamada, K.; Aoyama, Y.; Fukao, T.; Densupsoontorn, N.; Jirapinyo, P.; Sathienkijkanchai, A.; Yamaguchi, S.; Wasant, P. Carnitine-acylcarnitine translocase deficiency: Two neonatal cases with common splicing mutation and in vitro bezafibrate response. Brain Dev. 2015, 37, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.W.; Lai, C.K.; Chow, C.B.; Tong, S.-F.; Yuen, Y.-P.; Mak, Y.-F.; Chan, Y.-W. Ethnic-specific splicing mutation of the carnitine-acylcarnitine translocase gene in a Chinese neonate presenting with sudden unexpected death. Chin. Med. J. 2003, 116, 1110–1112. [Google Scholar] [PubMed]
- Fukushima, T.; Kaneoka, H.; Yasuno, T.; Sasaguri, Y.; Tokuyasu, T.; Tokoro, K.; Fukao, T.; Saito, T. Three novel mutations in the carnitine-acylcarnitine translocase (CACT) gene in patients with CACT deficiency and in healthy individuals. J. Hum. Genet. 2013, 58, 788–793. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| µmol/L | At 24 h of Life |
|---|---|
| C0 (≥10.85) | 9.45 |
| C2 (≥3.35) | 12.67 |
| C14 (≤0.58) | 1.74 |
| C16 (≤6.22) | 19.98 |
| C18 (≤1.87) | 3.75 |
| C10 (≤0.4) | 0.98 |
| C12 (≤0.4) | 1.27 |
| (µmol/L) | At 30 h of Life |
|---|---|
| C16 (<7) | 15.06 |
| C18 (<2.3) | 2.75 |
| C18:2 (<0.98) | 0.20 |
| C16:1 (<0.55) | 1.21 |
| CPT2 (<27.85) | 0.29 |
| C18/C3 (≤2.15) | 4.63 |
| C18:1/C8 (≤66) | 24.89 |
| C0/(C16+C18) (>1.34) | 0.37 |
| C18:1 (<3.04) | 3.49 |
| C0 (≥100) | 6.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona, S.M.G.; Abacan, M.A.R.; Alcausin, M.M.L.B. Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing. Int. J. Neonatal Screen. 2023, 9, 4. https://doi.org/10.3390/ijns9010004
Carmona SMG, Abacan MAR, Alcausin MMLB. Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing. International Journal of Neonatal Screening. 2023; 9(1):4. https://doi.org/10.3390/ijns9010004
Chicago/Turabian StyleCarmona, Suzanne Marie G., Mary Ann R. Abacan, and Maria Melanie Liberty B. Alcausin. 2023. "Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing" International Journal of Neonatal Screening 9, no. 1: 4. https://doi.org/10.3390/ijns9010004
APA StyleCarmona, S. M. G., Abacan, M. A. R., & Alcausin, M. M. L. B. (2023). Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing. International Journal of Neonatal Screening, 9(1), 4. https://doi.org/10.3390/ijns9010004