
Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing


Abstract
1. Introduction
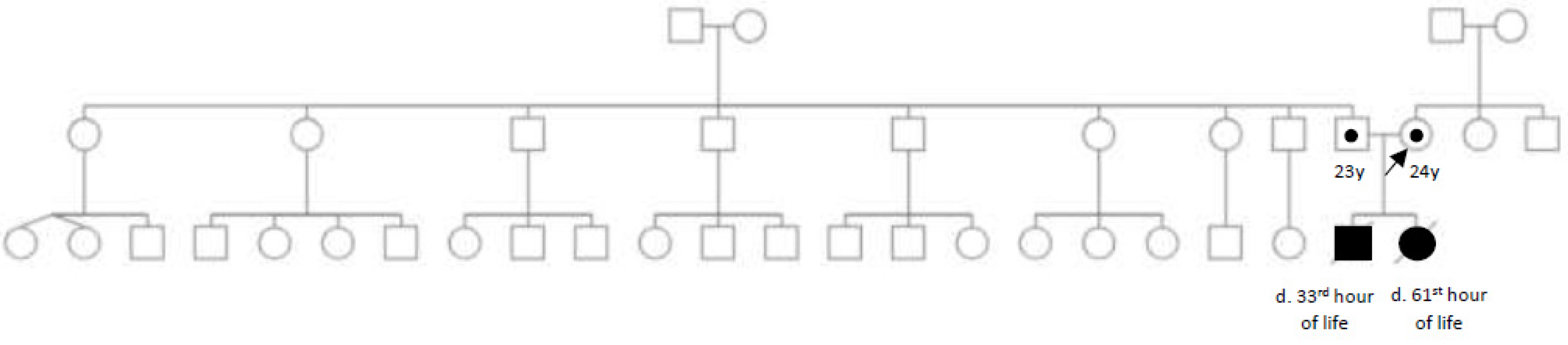
2. Case Report
3. Investigations
3.1. Biochemical Tests
3.2. Genetic Tests
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iacobazzi, V.; Invernizzi, F.; Baratta, S.; Pons, R.; Chung, W.; Garavaglia, B.; Dionisi-Vici, C.; Ribes, A.; Parini, R.; Dolores Huertas, M.; et al. Molecular and functional analysis of SLC25A20 mutations causing carnitine-acylcarnitine translocase deficiency. Hum. Mutat. 2004, 24, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Gozalbo, M.; Bakker, J.; Waterham, H.; Wanders, R. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol. Asp. Med. 2004, 25, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.-M.; Hu, H.; Ahmed, A.; Feng, B.-B.; Liu, J.; Jia, Z.-J.; Wang, H. Carnitine-acylcarnitine translocase deficiency with c.199-10 t>g and novel c.1a>g mutation. Medicine 2017, 96, e8549. [Google Scholar] [CrossRef] [PubMed]
- Vitoria, I.; Martín-Hernández, E.; Peña-Quintana, L.; Bueno, M.; Quijada-Fraile, P.; Dalmau, J.; Molina-Marrero, S.; Pérez, B.; Merinero, B. Carnitineacylcarnitine translocase deficiency: Experience with four cases in Spain and review of the literature. JIMD Rep. 2015, 20, 11–20. [Google Scholar] [PubMed]
- Hsu, B.Y.; Iacobazzi, V.; Wang, Z.; Harvie, H.; Chalmers, R.; Saudubray, J.; Palmieri, F.; Ganguly, A.; Stanley, C. Aberrant mRNA splicing associated with coding region mutations in children with carnitine-acylcarnitine translocase deficiency. Mol. Genet. Metab. 2001, 74, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Hoffmann, G.F.; Matern, D. Newborn screening for disorders of fatty-acid oxidation: Experience and recommendations from an expert meeting. J. Inherit. Metab. Dis. 2010, 33, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.; Tang, N.L.; Li, C.K.; Law, L.K.; To, K.F.; Yau, P.; Fung, S.L.M.; Chong, J.S.C.; Tsung, L.; Chiang, G.; et al. Inherited metabolic diseases in the Southern Chinese population: Spectrum of diseases and estimated incidence from recurrent mutations. Pathology 2014, 46, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Cai, Y.; Li, S.; Xiong, H.; Liu, M.; Ma, F.; Xiao, X.; Hao, H. Late-Onset Carnitine–Acylcarnitine Translocase Deficiency with SLC25A20 c.199-10T>G Variation: Case Report and Pathologic Analysis of Liver Biopsy. Front. Pediatr. 2020, 8, 585646. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.S.; Lam, C.W.; Lai, C.K.; Yuen, Y.P.; Chan, K.Y.; Shek, C.C.; Chan, A.Y.W.; Chow, C.B. Carnitineacylcarnitine translocase deficiency in three neonates presenting with rapid deterioration and cardiac arrest. Hong Kong Med. J. 2007, 13, 66–68. [Google Scholar] [PubMed]
- Hoffmann, G.I.; Nyhan, W.L.; Zschacke, J.; Kahler, S.G.; Mayatepek, E. (Eds.) Inherited Metabolic Diseases, 1st ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2002; p. 82. [Google Scholar]
- Costa, C.; Costa, J.M.; Slama, A.; Boutron, A.; Vequaud, C.; Legrand, A.; Brivet, M. Mutational spectrum and DNA-based prenatal diagnosis in carnitine-acyl carnitine translocate deficiency. Mol. Genet. Metab. 2003, 78, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Vatanavicharn, N.; Yamada, K.; Aoyama, Y.; Fukao, T.; Densupsoontorn, N.; Jirapinyo, P.; Sathienkijkanchai, A.; Yamaguchi, S.; Wasant, P. Carnitine-acylcarnitine translocase deficiency: Two neonatal cases with common splicing mutation and in vitro bezafibrate response. Brain Dev. 2015, 37, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.W.; Lai, C.K.; Chow, C.B.; Tong, S.-F.; Yuen, Y.-P.; Mak, Y.-F.; Chan, Y.-W. Ethnic-specific splicing mutation of the carnitine-acylcarnitine translocase gene in a Chinese neonate presenting with sudden unexpected death. Chin. Med. J. 2003, 116, 1110–1112. [Google Scholar] [PubMed]
- Fukushima, T.; Kaneoka, H.; Yasuno, T.; Sasaguri, Y.; Tokuyasu, T.; Tokoro, K.; Fukao, T.; Saito, T. Three novel mutations in the carnitine-acylcarnitine translocase (CACT) gene in patients with CACT deficiency and in healthy individuals. J. Hum. Genet. 2013, 58, 788–793. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| µmol/L | At 24 h of Life |
|---|---|
| C0 (≥10.85) | 9.45 |
| C2 (≥3.35) | 12.67 |
| C14 (≤0.58) | 1.74 |
| C16 (≤6.22) | 19.98 |
| C18 (≤1.87) | 3.75 |
| C10 (≤0.4) | 0.98 |
| C12 (≤0.4) | 1.27 |
| (µmol/L) | At 30 h of Life |
|---|---|
| C16 (<7) | 15.06 |
| C18 (<2.3) | 2.75 |
| C18:2 (<0.98) | 0.20 |
| C16:1 (<0.55) | 1.21 |
| CPT2 (<27.85) | 0.29 |
| C18/C3 (≤2.15) | 4.63 |
| C18:1/C8 (≤66) | 24.89 |
| C0/(C16+C18) (>1.34) | 0.37 |
| C18:1 (<3.04) | 3.49 |
| C0 (≥100) | 6.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona, S.M.G.; Abacan, M.A.R.; Alcausin, M.M.L.B. Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing. Int. J. Neonatal Screen. 2023, 9, 4. https://doi.org/10.3390/ijns9010004
Carmona SMG, Abacan MAR, Alcausin MMLB. Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing. International Journal of Neonatal Screening. 2023; 9(1):4. https://doi.org/10.3390/ijns9010004
Chicago/Turabian StyleCarmona, Suzanne Marie G., Mary Ann R. Abacan, and Maria Melanie Liberty B. Alcausin. 2023. "Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing" International Journal of Neonatal Screening 9, no. 1: 4. https://doi.org/10.3390/ijns9010004
APA StyleCarmona, S. M. G., Abacan, M. A. R., & Alcausin, M. M. L. B. (2023). Carnitine-acylcarnitine Translocase Deficiency with c.199-10T>G Mutation in Two Filipino Neonates Detected through Parental Carrier Testing. International Journal of Neonatal Screening, 9(1), 4. https://doi.org/10.3390/ijns9010004