Migraine Visual Aura and Cortical Spreading Depression—Linking Mathematical Models to Empirical Evidence




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
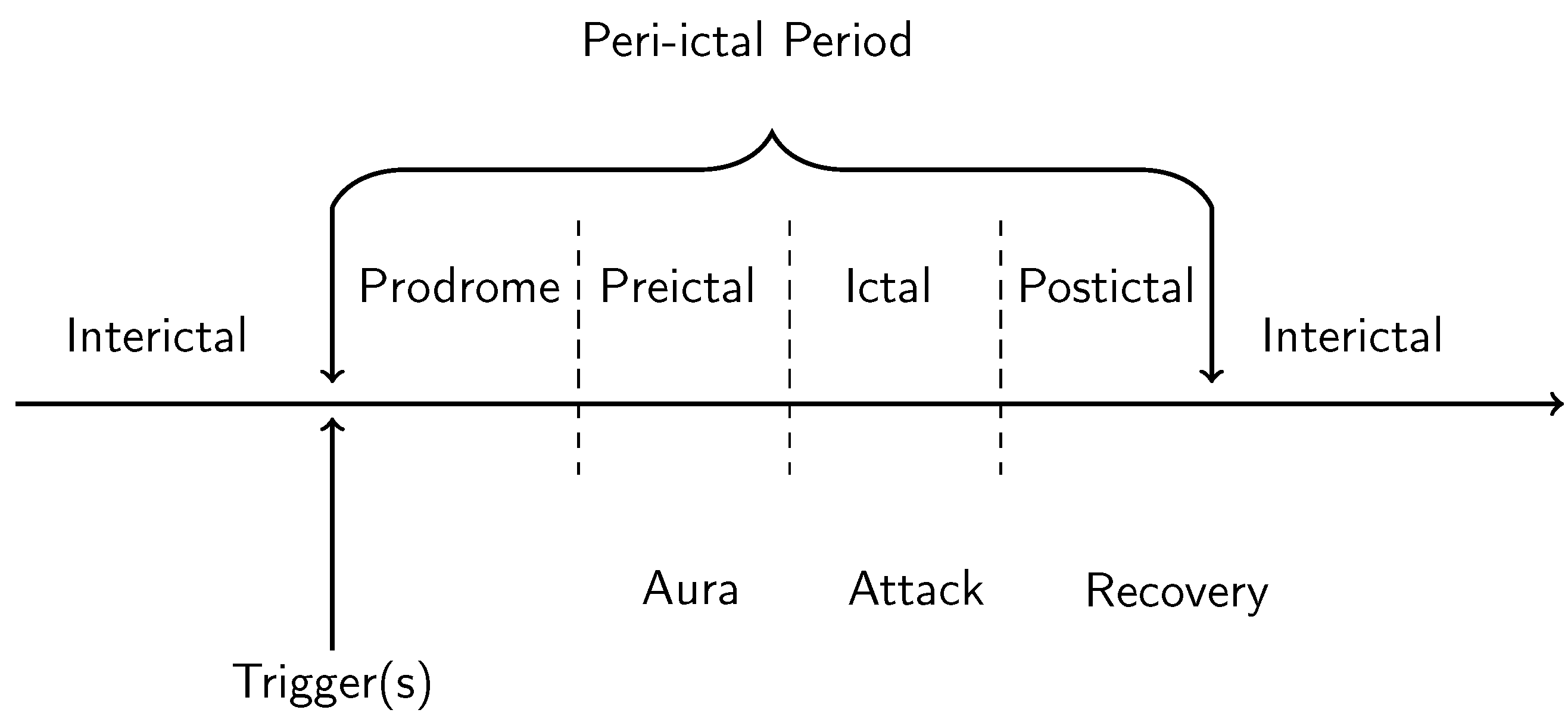
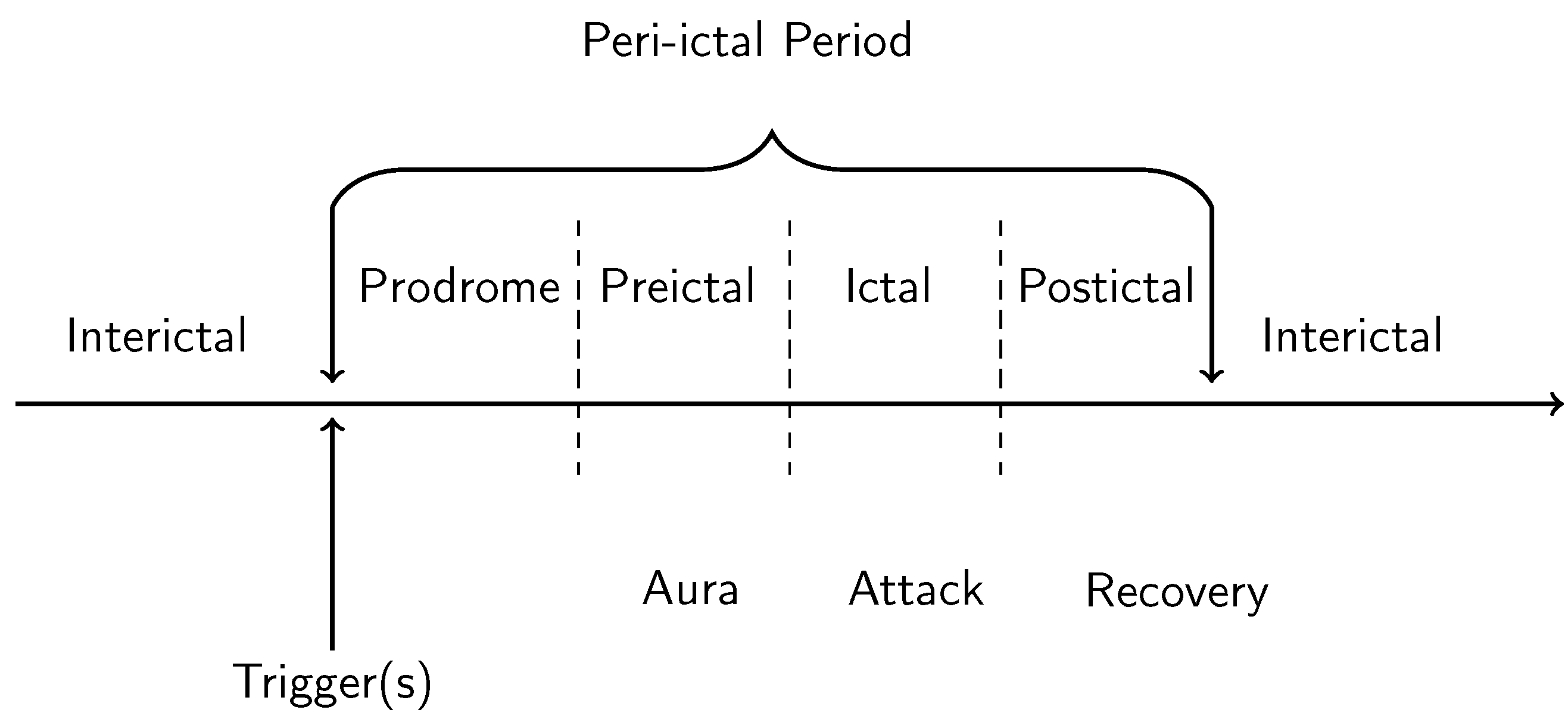
1.1. Phenomenology of Hallucinations and Visual Aura
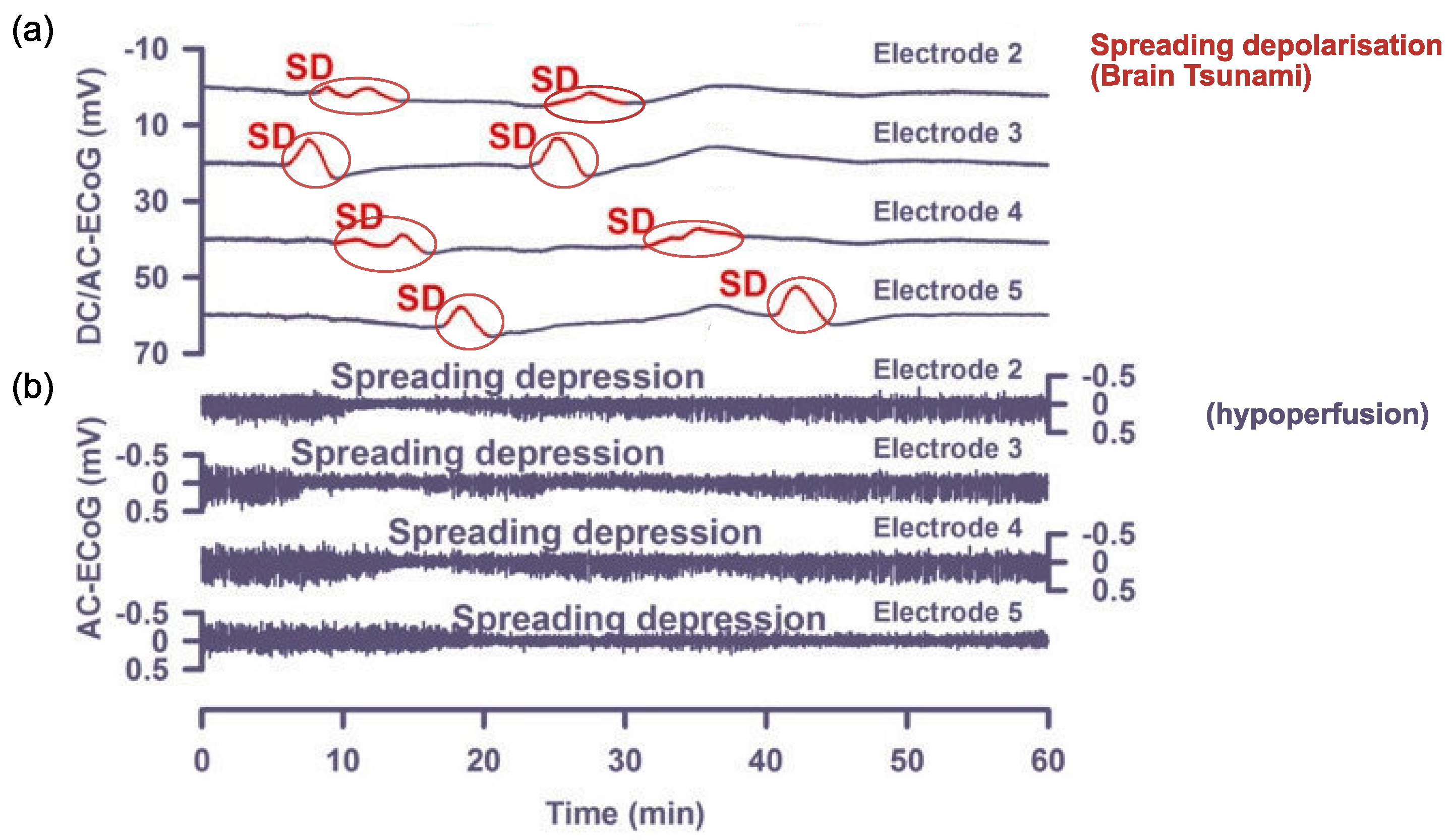
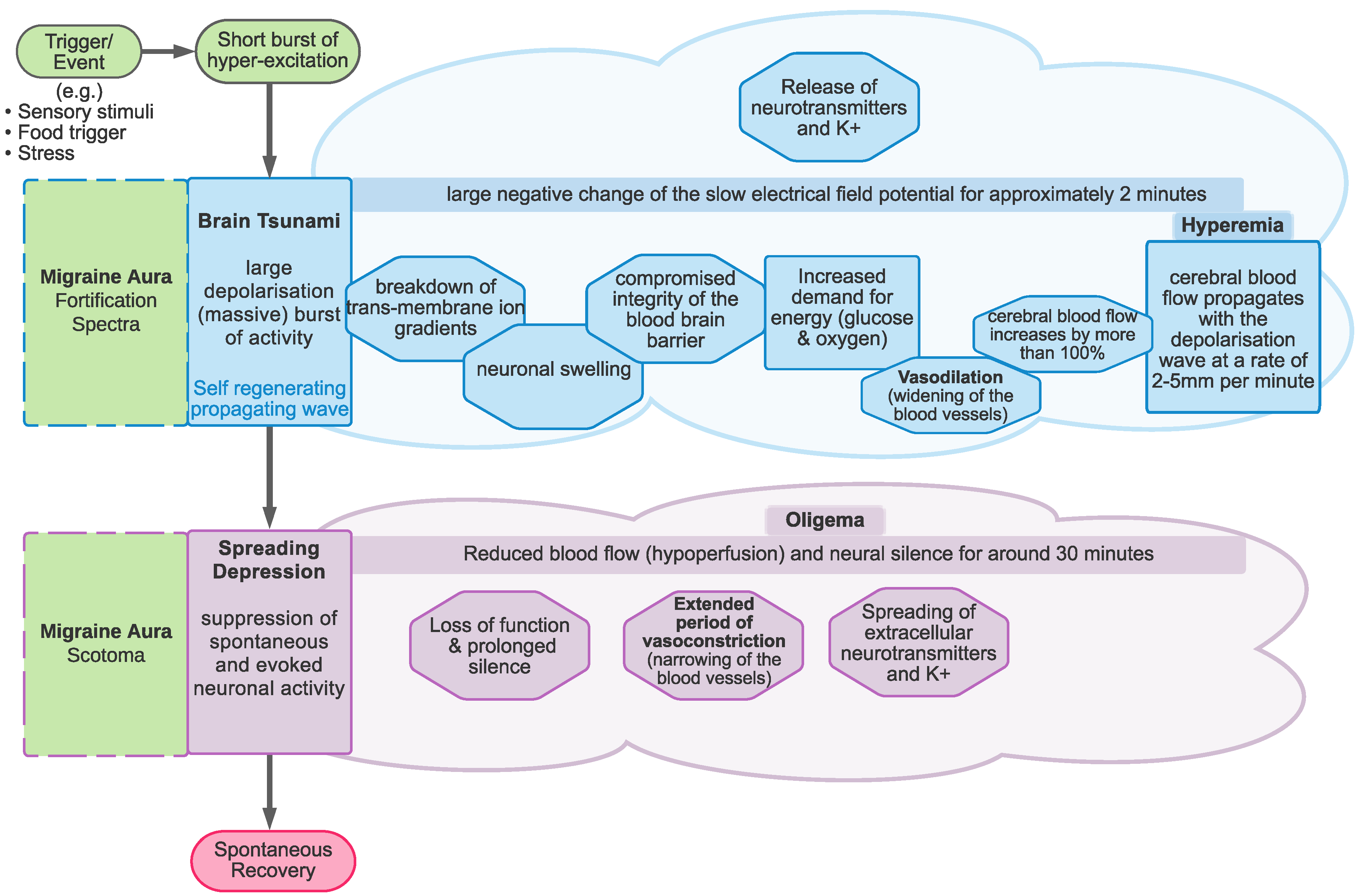
1.2. Cortical Spreading Depression as the Proposed Physiological Correlate of Aura
2. Models of Cortical Spreading Depression
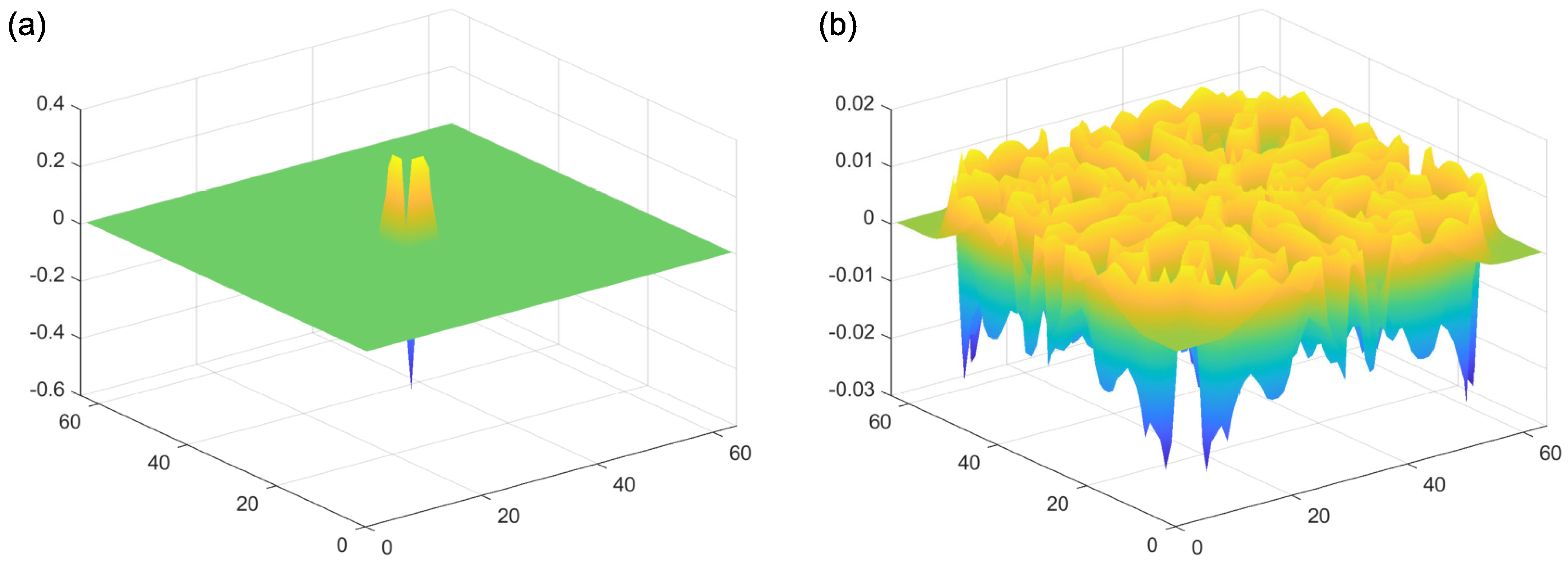
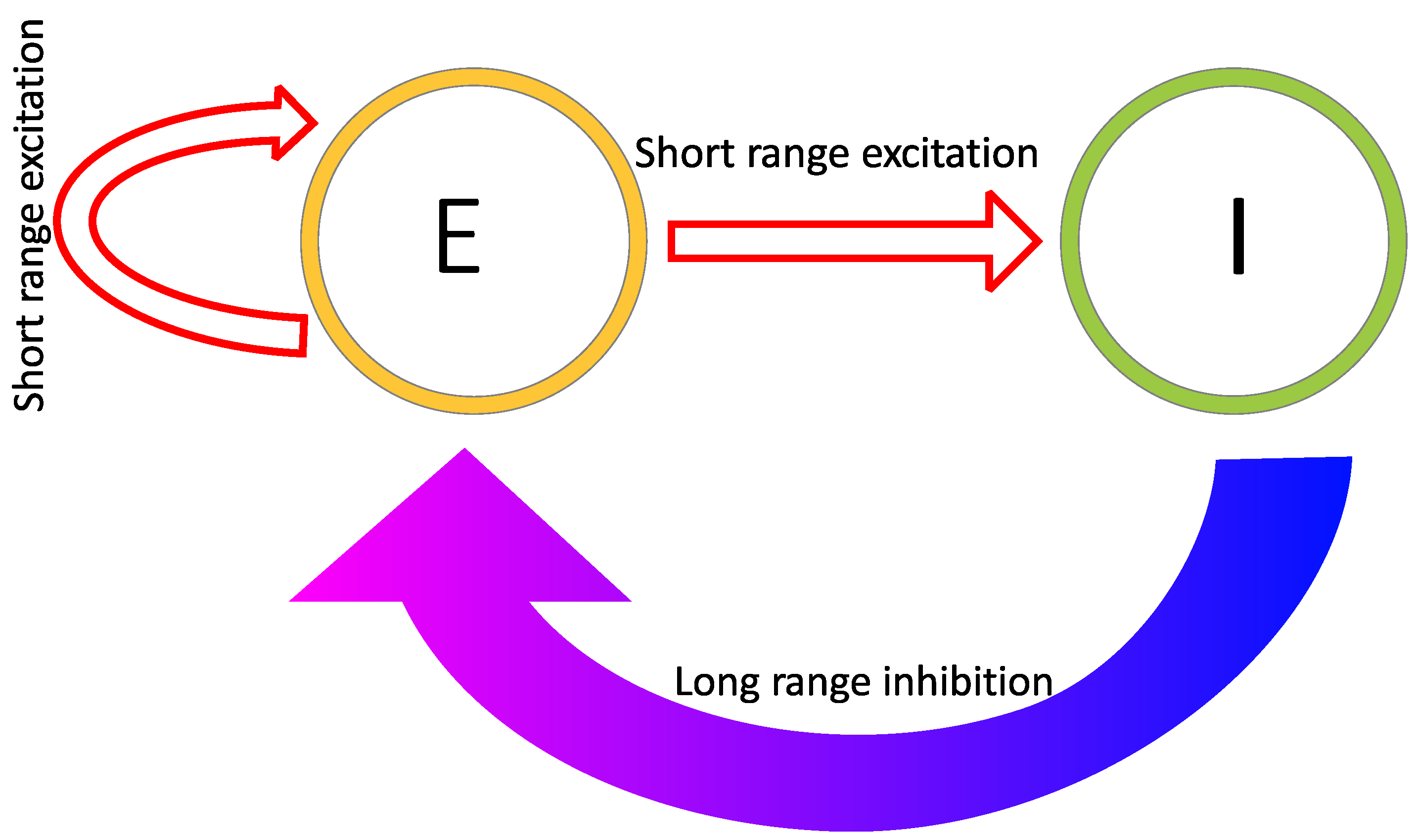
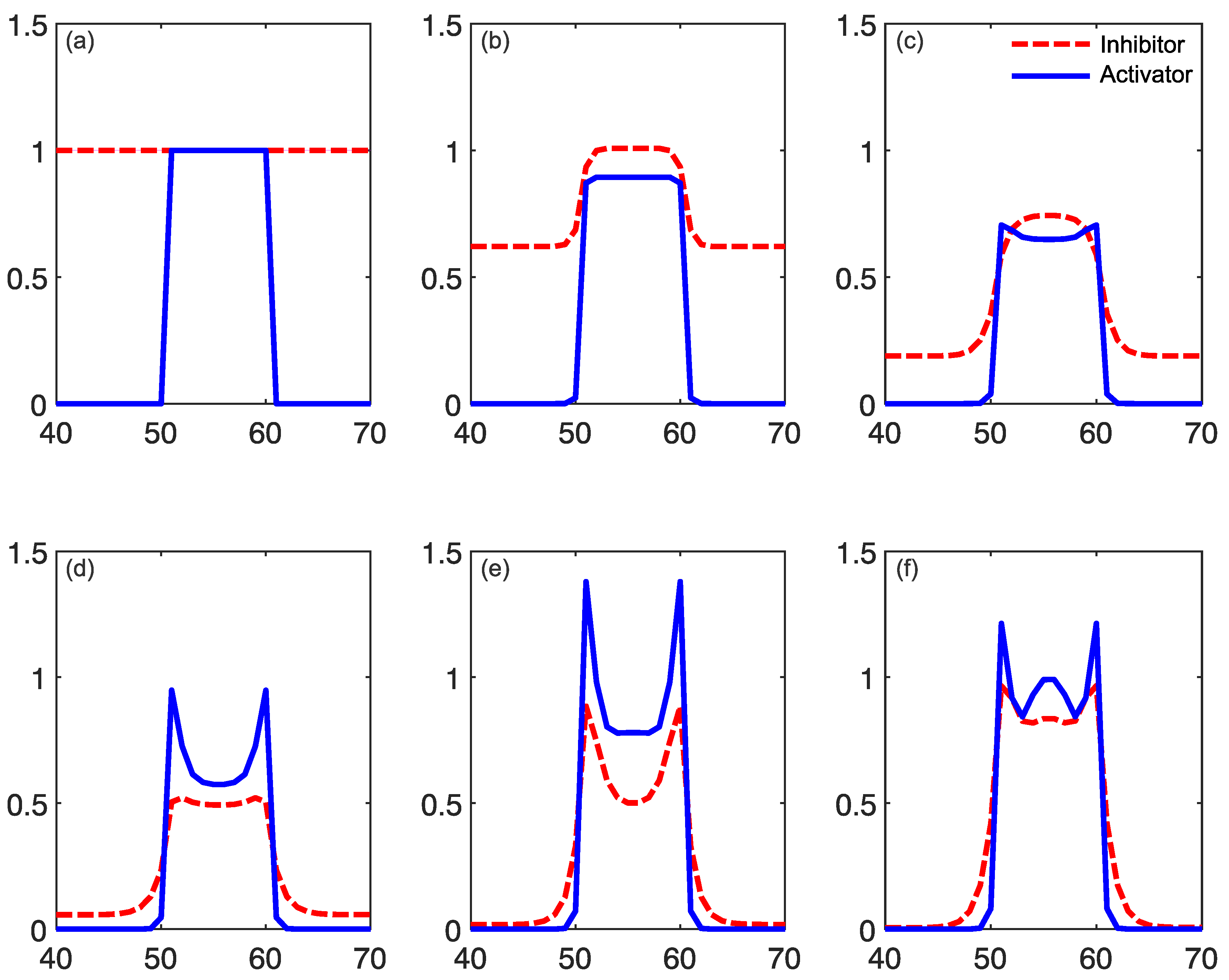
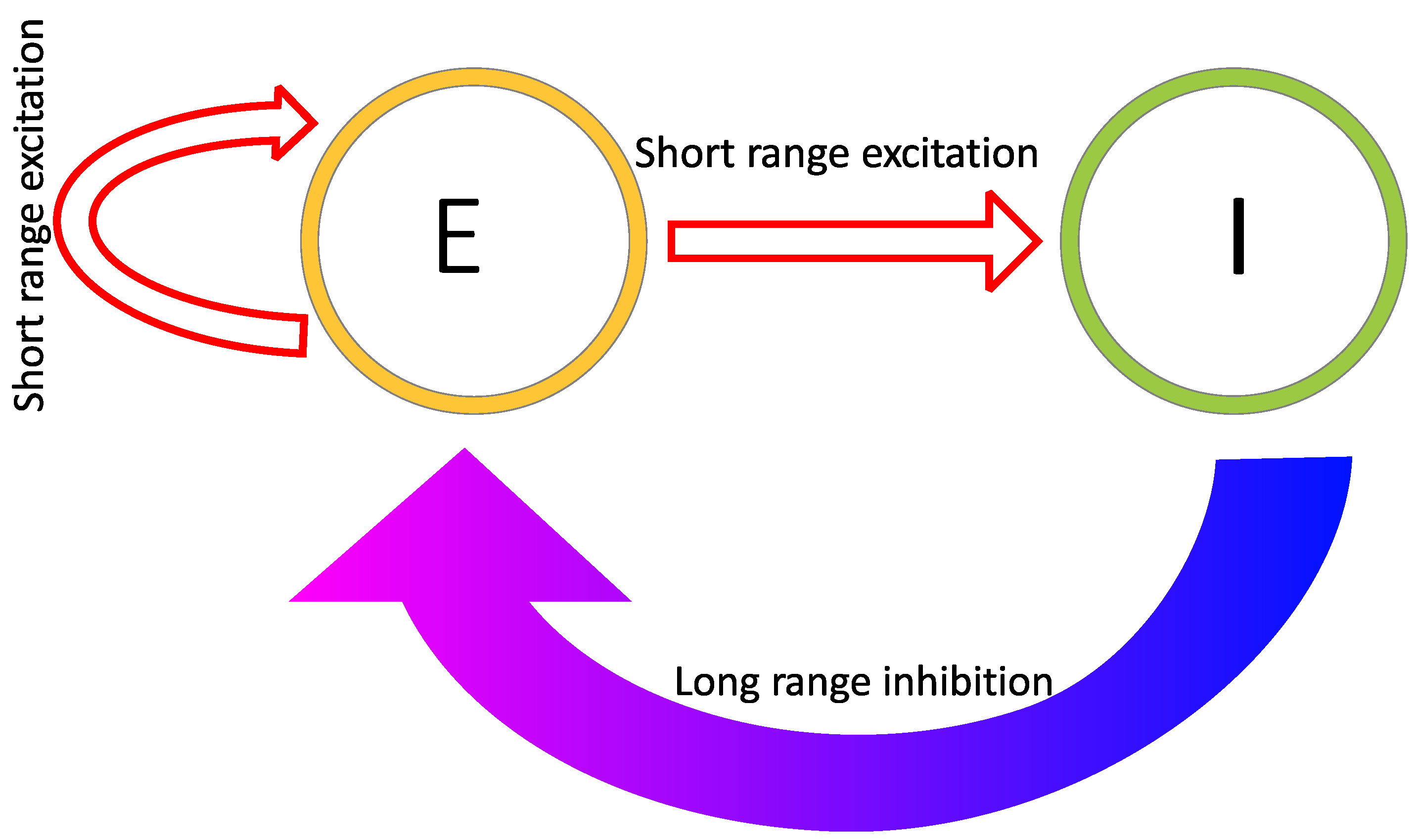
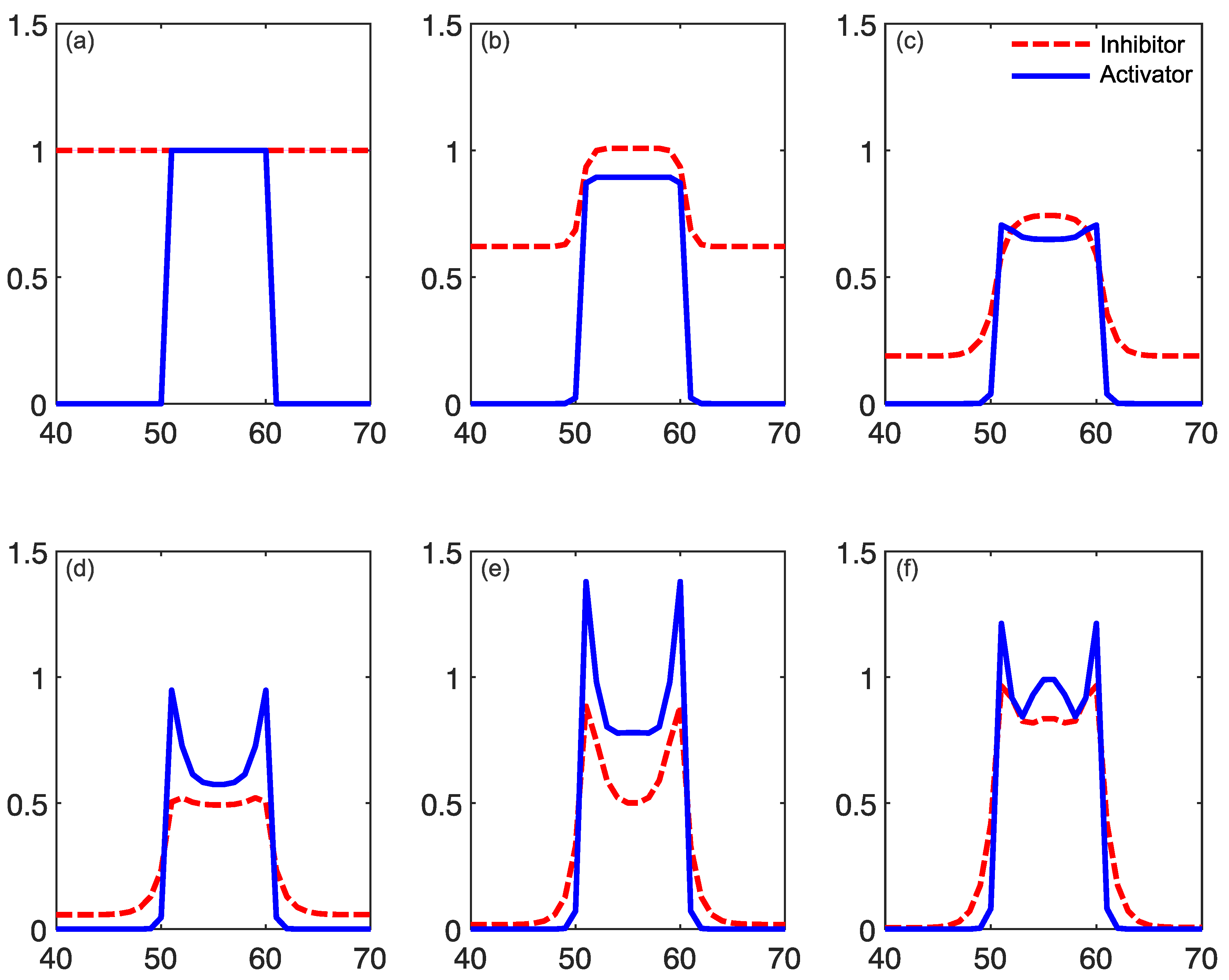
2.1. Reaction–Diffusion Models
2.2. Reaction–Diffusion Models and the Brain
2.3. What Do Models Account for?
3. Linking Models to Physiological Differences in Migraine
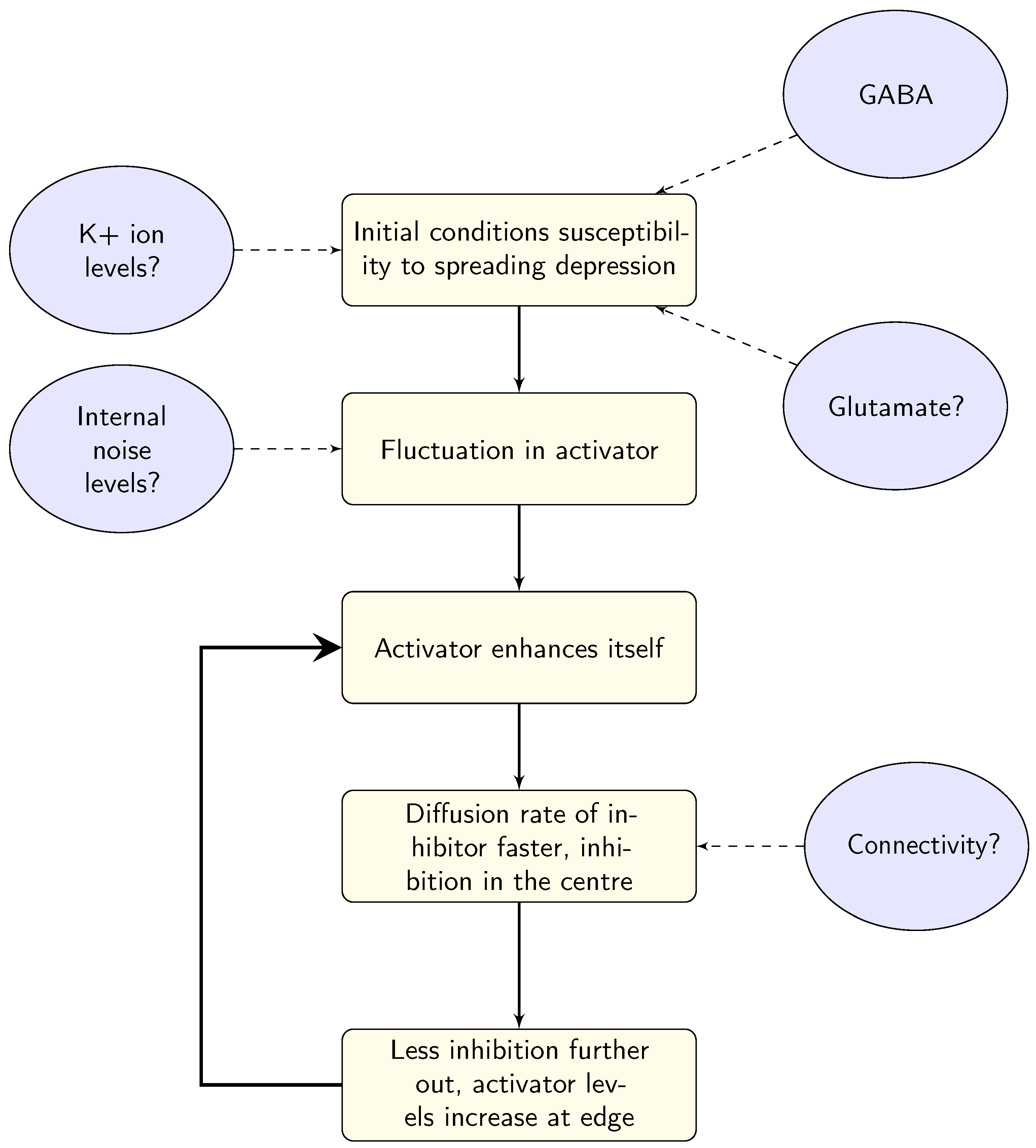
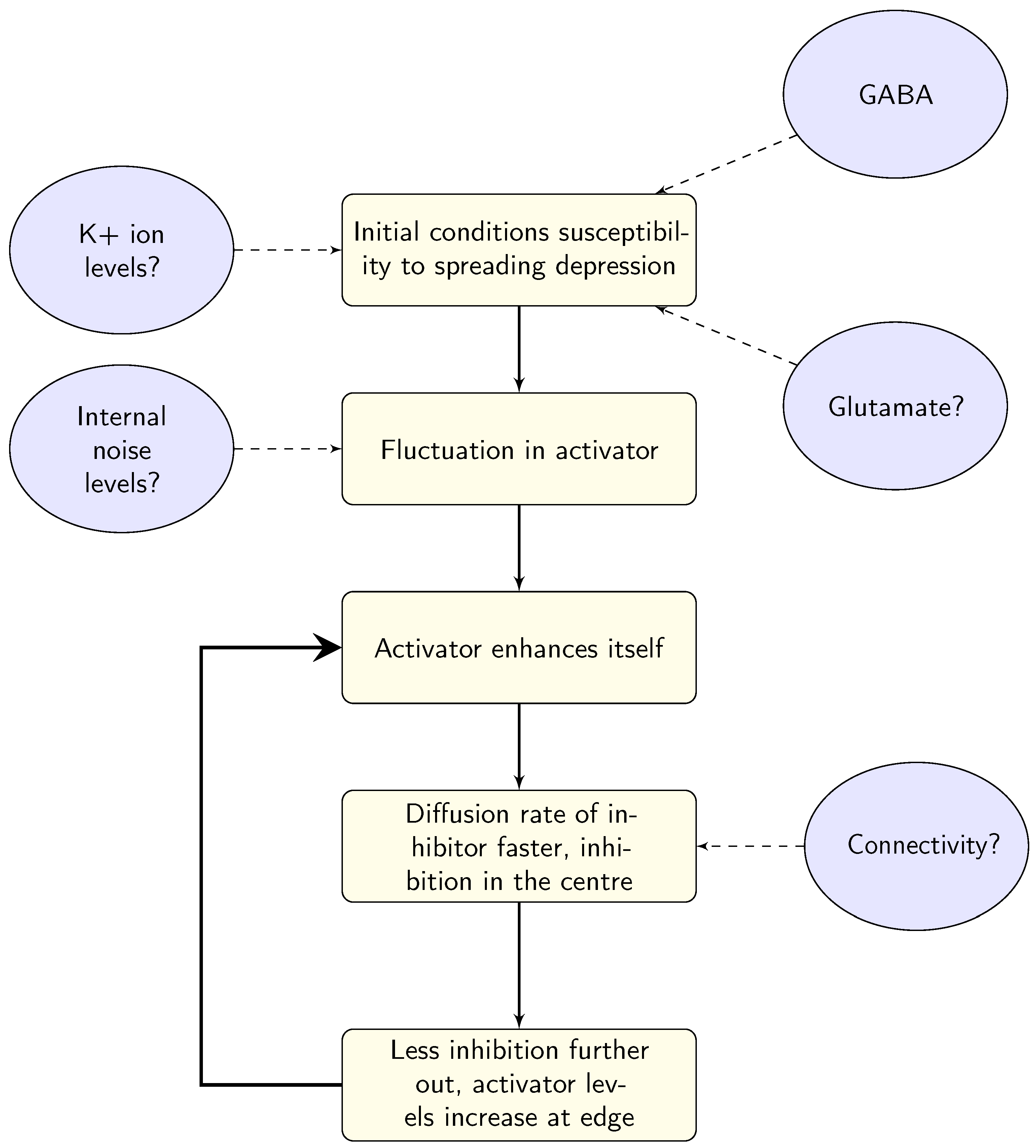
3.1. Ion Transfer
3.2. GABA
3.3. Glutamate
3.4. Electrical Stimulation
3.5. Links between Models and Biology
4. Linking Models to Known Migraine Triggers
4.1. Food-Based Triggers
4.2. Sensory Triggers
4.3. Modelling Triggers
5. Linking Models to Neural Activity during Aura (and Headache)
5.1. Slow Waves
5.2. Beta Band Oscillations
5.3. Alpha Band Oscillations
5.4. Linking Electrophysiology to Reaction–Diffusion Models—Oscillations
5.5. Linking Electrophysiology to Models—Connectivity
6. Linking Models to Psychophysical Evidence and Signal Detection Models
6.1. Differences in Performance over the Migraine Cycle
6.2. Linking Back to the Models
7. Discussion and Unresolved Questions
7.1. Arguments against Cortical Spreading Depression as the Mechanism for Migraine
7.2. Can We Infer Clues to Aura Susceptibility from the Models?
7.3. How the Models Account for Pain
7.4. Long-Term Effects and Stroke Risk
7.5. Age and Sex Differences
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AC | Alternating current |
| BAM | Basilar-type-migraine |
| DC | Direct current |
| GABA | Gamma-aminobutyric acid |
| Glx | Combined glutamate and glutamine complex |
| K+ | Potassium ion |
| MA | Migraine with aura |
| MEG | Magnetoencephalogram |
| MO | Migraine without aura |
| MR | Magnetic resonance |
| MT | Middle-temporal |
| NDMA | Nitrosodimethylamine |
| PSD | Power spectral density |
| rCBF | Cerebral blood flow |
| tACS | Transcranial alternating current stimulation |
| tDCS | Transcranial direct current stimulation |
| TMS | Transcranial magnetic stimulation |
| Xe | Xenon |
References
- Kelman, L. The aura: A tertiary care study of 952 migraine patients. Cephalalgia 2004, 24, 728–734. [Google Scholar] [CrossRef]
- Smith, J.M.; Bradley, D.P.; James, M.F.; Huang, C.L.H. Physiological studies of cortical spreading depression. Biol. Rev. 2006, 81, 457–481. [Google Scholar] [CrossRef]
- Teeple, R.C.; Caplan, J.P.; Stern, T.A. Visual hallucinations: Differential diagnosis and treatment. Prim. Care Companion J. Clin. Psychiatry 2009, 11, 26. [Google Scholar] [CrossRef]
- Steiner, T.; Scher, A.; Stewart, W.; Kolodner, K.; Liberman, J.; Lipton, R. The prevalence and disability burden of adult migraine in England and their relationships to age, gender and ethnicity. Cephalalgia 2003, 23, 519–527. [Google Scholar] [CrossRef]
- Viana, M.; Sances, G.; Linde, M.; Ghiotto, N.; Guaschino, E.; Allena, M.; Terrazzino, S.; Nappi, G.; Goadsby, P.J.; Tassorelli, C. Clinical features of migraine aura: Results from a prospective diary-aided study. Cephalalgia 2017, 37, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.; Sprenger, T.; Andelova, M.; Goadsby, P.J. The typical duration of migraine aura: A systematic review. Cephalalgia 2013, 33, 483–490. [Google Scholar] [CrossRef]
- Viana, M.; Sances, G.; Linde, M.; Nappi, G.; Khaliq, F.; Goadsby, P.J.; Tassorelli, C. Prolonged migraine aura: New insights from a prospective diary-aided study. J. Headache Pain 2018, 19, 1–5. [Google Scholar] [CrossRef]
- Queiroz, L.P.; Friedman, D.I.; Rapoport, A.M.; Purdy, R.A. Characteristics of migraine visual aura in Southern Brazil and Northern USA. Cephalalgia 2011, 31, 1652–1658. [Google Scholar] [CrossRef]
- Viana, M.; Tronvik, E.A.; Do, T.P.; Zecca, C.; Hougaard, A. Clinical features of visual migraine aura: A systematic review. J. Headache Pain 2019, 20, 64. [Google Scholar] [CrossRef] [Green Version]
- Plant, G.T. The fortification spectra of migraine. Br. Med. J. Clin. Res. Ed. 1986, 293, 1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, O. Migraine; Vintage Books: Berkeley, CA, USA, 1992. [Google Scholar]
- Schott, G. Exploring the visual hallucinations of migraine aura: The tacit contribution of illustration. Brain 2007, 130, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Lashley, K.S. Patterns of cerebral integration indicated by the scotomas of migraine. Arch. Neurol. Psychiatry 1941, 46, 331–339. [Google Scholar] [CrossRef]
- Panayiotopoulos, C. Elementary visual hallucinations in migraine and epilepsy. J. Neurol. Neurosurg. Psychiatry 1994, 57, 1371–1374. [Google Scholar] [CrossRef] [Green Version]
- Martínez, J.V.L.; Specialli, J.G. Migraine with visual aura versus occipital epilepsy. Headache J. Head Face Pain Lett. Ed. 1997, 37, 113. [Google Scholar] [CrossRef]
- Panayiotopoulos, C. Elementary visual hallucinations, blindness, and headache in idiopathic occipital epilepsy: Differentiation from migraine. J. Neurol. Neurosurg. Psychiatry 1999, 66, 536–540. [Google Scholar] [CrossRef]
- Panayiotopoulos, C.P. Visual aura of migraine versus visual occipital lobe seizures. Cephalalgia 2012, 32, 654. [Google Scholar] [CrossRef]
- Sturzenegger, M.H.; Meienberg, O. Basilar artery migraine: A follow-up study of 82 cases. Headache J. Head Face Pain 1985, 25, 408–415. [Google Scholar] [CrossRef]
- Siegel, R.K. Cocaine hallucinations. Am. J. Psychiatry 1978, 135, 309–314. [Google Scholar] [PubMed]
- Dybowski, M. Conditions for the Appearance of Hypnagogic Visions; Poznanskie Towarzystwo Psychologiozne: Poznań, Poland, 1939. [Google Scholar]
- Ermentrout, G.B.; Cowan, J.D. A mathematical theory of visual hallucination patterns. Biol. Cybern. 1979, 34, 137–150. [Google Scholar] [CrossRef]
- Eriksen, M.; Thomsen, L.; Andersen, I.; Nazim, F.; Olesen, J. Clinical characteristics of 362 patients with familial migraine with aura. Cephalalgia 2004, 24, 564–575. [Google Scholar] [CrossRef]
- Hansen, J.M.; Baca, S.M.; VanValkenburgh, P.; Charles, A. Distinctive anatomical and physiological features of migraine aura revealed by 18 years of recording. Brain 2013, 136, 3589–3595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowyer, S.; Tepley, N.; Papuashvili, N.A.; Kato, S.; Barkley, G.; Welch, K.; Okada, Y. Analysis of MEG signals of spreading cortical depression with propagation constrained to a rectangular cortical strip: II. Gyrencephalic swine model. Brain Res. 1999, 843, 79–86. [Google Scholar] [CrossRef]
- Hadjikhani, N.; Del Rio, M.S.; Wu, O.; Schwartz, D.; Bakker, D.; Fischl, B.; Kwong, K.K.; Cutrer, F.M.; Rosen, B.R.; Tootell, R.B.; et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc. Natl. Acad. Sci. USA 2001, 98, 4687–4692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, J.P. Spreading depolarization, Tsunami im Hirn. e-Neuroforum 2009, 15, 108–113. [Google Scholar] [CrossRef]
- Dreier, J.P.; Major, S.; Foreman, B.; Winkler, M.K.; Kang, E.J.; Milakara, D.; Lemale, C.L.; DiNapoli, V.; Hinzman, J.M.; Woitzik, J.; et al. Terminal spreading depolarization and electrical silence in death of human cerebral cortex. Ann. Neurol. 2018, 83, 295–310. [Google Scholar] [CrossRef] [Green Version]
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of migraine: A disorder of sensory processing. Physiol. Rev. 2017, 97, 553–622. [Google Scholar] [CrossRef]
- Leo, A.A. Pial circulation and spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1944, 7, 391–396. [Google Scholar] [CrossRef]
- Leao, A.A. Spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1944, 7, 359–390. [Google Scholar] [CrossRef]
- Lauritzen, M.; Dreier, J.P.; Fabricius, M.; Hartings, J.A.; Graf, R.; Strong, A.J. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J. Cereb. Blood Flow Metab. 2011, 31, 17–35. [Google Scholar] [CrossRef]
- Wilkinson, F. Auras and other hallucinations: Windows on the visual brain. Prog. Brain Res. 2004, 144, 305–320. [Google Scholar]
- Dahlem, M.A.; Müller, S. Migraine aura dynamics after reverse retinotopic mapping of weak excitation waves in the primary visual cortex. Biol. Cybern. 2003, 88, 419–424. [Google Scholar] [CrossRef]
- Charles, A.; Brennan, K. Cortical spreading depression—New insights and persistent questions. Cephalalgia 2009, 29, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Hartings, J.A.; Wilson, J.A.; Hinzman, J.M.; Pollandt, S.; Dreier, J.P.; DiNapoli, V.; Ficker, D.M.; Shutter, L.A.; Andaluz, N. Spreading depression in continuous electroencephalography of brain trauma. Ann. Neurol. 2014, 76, 681–694. [Google Scholar] [CrossRef]
- Charles, A.C.; Baca, S.M. Cortical spreading depression and migraine. Nat. Rev. Neurol. 2013, 9, 637. [Google Scholar] [CrossRef]
- Hofmeijer, J.; Van Kaam, C.; van de Werff, B.; Vermeer, S.E.; Tjepkema-Cloostermans, M.C.; van Putten, M.J. Detecting cortical spreading depolarization with full band scalp electroencephalography: An illusion? Front. Neurol. 2018, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Harriott, A.M.; Takizawa, T.; Chung, D.Y.; Chen, S.P. Spreading depression as a preclinical model of migraine. J. Headache Pain 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Billock, V.A.; Tsou, B.H. Elementary visual hallucinations and their relationships to neural pattern-forming mechanisms. Psychol. Bull. 2012, 138, 744. [Google Scholar] [CrossRef] [Green Version]
- Zandt, B.J.; ten Haken, B.; van Putten, M.J.; Dahlem, M.A. How does spreading depression spread? Physiology and modeling. Rev. Neurosci. 2015, 26, 183–198. [Google Scholar] [CrossRef] [Green Version]
- Dahlem, M.A.; Hadjikhani, N. Migraine aura: Retracting particle-like waves in weakly susceptible cortex. PLoS ONE 2009, 4, e5007. [Google Scholar] [CrossRef]
- Dahlem, M.A.; Müller, S.C. Self-induced splitting of spiral-shaped spreading depression waves in chicken retina. Exp. Brain Res. 1997, 115, 319–324. [Google Scholar] [CrossRef]
- Wilson, H.R.; Cowan, J.D. Excitatory and inhibitory interactions in localized populations of model neurons. Biophys. J. 1972, 12, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Wilson, H.R.; Cowan, J.D. A mathematical theory of the functional dynamics of cortical and thalamic nervous tissue. Kybernetik 1973, 13, 55–80. [Google Scholar] [CrossRef]
- Turing, A. The chemical basis of morphogenesis. Philos. Trans. R. Soc. Lond. 1952, B237. [Google Scholar] [CrossRef]
- Kondo, S.; Miura, T. Reaction-diffusion model as a framework for understanding biological pattern formation. Science 2010, 329, 1616–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, P.; Scott, S.K. Chemical Oscillations and Instabilities: Non-Linear Chemical Kinetics; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Garrity, M. How the Tiger Got Its Stripes. Available online: https://blogs.mathworks.com/graphics/2015/03/16/how-the-tiger-got-its-stripes/ (accessed on 27 February 2021).
- Gierer, A.; Meinhardt, H. A theory of biological pattern formation. Kybernetik 1972, 12, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhaoping, L.; Li, Z. Understanding Vision: Theory, Models, and Data; Oxford University Press: New York, NY, USA, 2014. [Google Scholar]
- Tass, P. Oscillatory Cortical Activity during Visual Hallucinations. J. Biol. Phys. 1997, 23, 21–66. [Google Scholar] [CrossRef]
- Reggia, J.A.; Montgomery, D. A computational model of visual hallucinations in migraine. Comput. Biol. Med. 1996, 26, 133–141. [Google Scholar] [CrossRef]
- Dahlem, M.; Engelmann, R.; Löwel, S.; Müller, S. Does the migraine aura reflect cortical organization? Eur. J. Neurosci. 2000, 12, 767–770. [Google Scholar] [CrossRef]
- Dahlem, M.; Chronicle, E. A computational perspective on migraine aura. Prog. Neurobiol. 2004, 74, 351–361. [Google Scholar] [CrossRef]
- Dahlem, M.A.; Müller, S.C. Reaction-diffusion waves in neuronal tissue and the window of cortical excitability. Ann. Phys. 2004, 13, 442–449. [Google Scholar] [CrossRef]
- Field, D.J.; Hayes, A.; Hess, R.F. Contour integration by the human visual system: Evidence for a local “association field”. Vis. Res. 1993, 33, 173–193. [Google Scholar] [CrossRef]
- Blasdel, G.; Obermayer, K.; Kiorpes, L. Organization of ocular dominance and orientation columns in the striate cortex of neonatal macaque monkeys. Vis. Neurosci. 1995, 12, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Daugman, J.G. Uncertainty relation for resolution in space, spatial frequency, and orientation optimized by two-dimensional visual cortical filters. JOSA A 1985, 2, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Field, D.J. What is the goal of sensory coding? Neural Comput. 1994, 6, 559–601. [Google Scholar] [CrossRef]
- Rule, M.; Stoffregen, M.; Ermentrout, B. A model for the origin and properties of flicker-induced geometric phosphenes. PLoS Comput. Biol. 2011, 7, e1002158. [Google Scholar] [CrossRef] [Green Version]
- Atasoy, S.; Donnelly, I.; Pearson, J. Human brain networks function in connectome-specific harmonic waves. Nat. Commun. 2016, 7, 10340. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.S.M.; Bhaumik, B. A reaction-diffusion model to capture disparity selectivity in primary visual cortex. PLoS ONE 2011, 6, e24997. [Google Scholar] [CrossRef] [PubMed]
- Seriès, P.; Latham, P.E.; Pouget, A. Tuning curve sharpening for orientation selectivity: Coding efficiency and the impact of correlations. Nat. Neurosci. 2004, 7, 1129–1135. [Google Scholar] [CrossRef]
- Dahlem, Y.A.; Dahlem, M.A.; Mair, T.; Braun, K.; Müller, S.C. Extracellular potassium alters frequency and profile of retinal spreading depression waves. Exp. Brain Res. 2003, 152, 221–228. [Google Scholar] [CrossRef]
- Hauge, A.W.; Kirchmann, M.; Olesen, J. Characterization of consistent triggers of migraine with aura. Cephalalgia 2011, 31, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Kilic, K.; Karatas, H.; Dönmez-Demir, B.; Eren-Kocak, E.; Gursoy-Ozdemir, Y.; Can, A.; Petit, J.M.; Magistretti, P.J.; Dalkara, T. Inadequate brain glycogen or sleep increases spreading depression susceptibility. Ann. Neurol. 2018, 83, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R.; Nicholas, E.; Petersen, N.C.; Dietz, A.G.; Xu, Q.; Sun, Q.; Nedergaard, M. Cortex-wide changes in extracellular potassium ions parallel brain state transitions in awake behaving mice. Cell Rep. 2019, 28, 1182–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, J.; Bes, A.; Kunkel, R.; Lance, J.W.; Nappi, G.; Pfaffenrath, V.; Rose, F.C.; Schoenberg, B.S.; Soyka, D.; Tfelt-Hansen, P.; et al. The international classification of headache disorders, (beta version). Cephalalgia 2013, 33, 629–808. [Google Scholar]
- Hounsgaard, J.; Nicholson, C. Potassium accumulation around individual purkinje cells in cerebellar slices from the guinea-pig. J. Physiol. 1983, 340, 359–388. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Chen, Y. Importance of astrocytes for potassium ion (K+) homeostasis in brain and glial effects of K+ and its transporters on learning. Neurosci. Biobehav. Rev. 2016, 71, 484–505. [Google Scholar] [CrossRef]
- Major, S.; Huo, S.; Lemale, C.L.; Siebert, E.; Milakara, D.; Woitzik, J.; Gertz, K.; Dreier, J.P. Direct electrophysiological evidence that spreading depolarization-induced spreading depression is the pathophysiological correlate of the migraine aura and a review of the spreading depolarization continuum of acute neuronal mass injury. GeroScience 2020, 42, 57–80. [Google Scholar] [CrossRef]
- Somjen, G.G. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev. 2001, 81, 1065–1096. [Google Scholar] [CrossRef] [Green Version]
- van den Maagdenberg, A.M.; Pietrobon, D.; Pizzorusso, T.; Kaja, S.; Broos, L.A.; Cesetti, T.; van de Ven, R.C.; Tottene, A.; van der Kaa, J.; Plomp, J.J.; et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 2004, 41, 701–710. [Google Scholar] [CrossRef]
- Turner, T.J.; Adams, M.E.; Dunlap, K. Calcium channels coupled to glutamate release identified by omega-Aga-IVA. Science 1992, 258, 310–313. [Google Scholar] [CrossRef]
- Timmermann, D.B.; Westenbroek, R.E.; Schousboe, A.; Catterall, W.A. Distribution of high-voltage-activated calcium channels in cultured γ-aminobutyric acidergic neurons from mouse cerebral cortex. J. Neurosci. Res. 2002, 67, 48–61. [Google Scholar] [CrossRef]
- Dietzel, I.; Heinemann, U.; Hofmeier, G.; Lux, H. Stimulus-induced changes in extracellular Na+ and Cl- concentration in relation to changes in the size of the extracellular space. Exp. Brain Res. 1982, 46, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Somjen, G.G. Na+ and K+ concentrations, extra-and intracellular voltages, and the effect of TTX in hypoxic rat hippocampal slices. J. Neurophysiol. 2000, 83, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Barolet, A.; Morris, M. Changes in extracellular K+ evoked by GABA, THIP and baclofen in the guinea-pig hippocampal slice. Exp. Brain Res. 1991, 84, 591–598. [Google Scholar] [CrossRef] [PubMed]
- DiNuzzo, M.; Mangia, S.; Maraviglia, B.; Giove, F. Physiological bases of the K+ and the glutamate/GABA hypotheses of epilepsy. Epilepsy Res. 2014, 108, 995–1012. [Google Scholar] [CrossRef] [Green Version]
- Van Harreveld, A.; Stamm, J. Cortical responses to metrazol and sensory stimulation in the rabbit. Electroencephalogr. Clin. Neurophysiol. 1955, 7, 363–370. [Google Scholar] [CrossRef]
- Palmer, J.; Chronicle, E.; Rolan, P.; Mulleners, W. Cortical hyperexcitability is cortical under-inhibition: Evidence from a novel functional test of migraine patients. Cephalalgia 2000, 20, 525–532. [Google Scholar] [CrossRef]
- Mulleners, W.M.; Chronicle, E.; Vredeveld, J.; Koehler, P. Visual cortex excitability in migraine before and after valproate prophylaxis: A pilot study using TMS. Eur. J. Neurol. 2002, 9, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Kurcyus, K.; Annac, E.; Hanning, N.M.; Harris, A.D.; Oeltzschner, G.; Edden, R.; Riedl, V. Opposite dynamics of GABA and glutamate levels in the occipital cortex during visual processing. J. Neurosci. 2018, 38, 9967–9976. [Google Scholar] [CrossRef]
- Dreier, J.P.; Major, S.; Pannek, H.W.; Woitzik, J.; Scheel, M.; Wiesenthal, D.; Martus, P.; Winkler, M.K.; Hartings, J.A.; Fabricius, M.; et al. Spreading convulsions, spreading depolarization and epileptogenesis in human cerebral cortex. Brain 2012, 135, 259–275. [Google Scholar] [CrossRef]
- Bigal, M.; Hetherington, H.; Pan, J.; Tsang, A.; Grosberg, B.; Avdievich, N.; Friedman, B.; Lipton, R. Occipital levels of GABA are related to severe headaches in migraine. Neurology 2008, 70, 2078–2080. [Google Scholar] [CrossRef] [Green Version]
- Stærmose, T.G.; Knudsen, M.K.; Kasch, H.; Blicher, J.U. Cortical GABA in migraine with aura-an ultrashort echo magnetic resonance spectroscopy study. J. Headache Pain 2019, 20, 110. [Google Scholar] [CrossRef]
- Bridge, H.; Stagg, C.J.; Near, J.; Lau, C.I.; Zisner, A.; Cader, M.Z. Altered neurochemical coupling in the occipital cortex in migraine with visual aura. Cephalalgia 2015, 35, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Aguila, M.E.R.; Rebbeck, T.; Leaver, A.M.; Lagopoulos, J.; Brennan, P.C.; Hübscher, M.; Refshauge, K.M. The association between clinical characteristics of migraine and brain GABA levels: An exploratory study. J. Pain 2016, 17, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, G.; Granella, F.; Cataldini, M.; Verdelli, F.; Balbi, T. GABA and glutamate in migraine. J. Headache Pain 2001, 2, s57–s60. [Google Scholar] [CrossRef] [Green Version]
- Chronicle, E.P.; Mulleners, W.M. Anticonvulsant drugs for migraine prophylaxis. Cochrane Database Syst. Rev. 2004, 3. [Google Scholar] [CrossRef]
- Cai, K.; Nanga, R.P.; Lamprou, L.; Schinstine, C.; Elliott, M.; Hariharan, H.; Reddy, R.; Epperson, C.N. The impact of gabapentin administration on brain GABA and glutamate concentrations: A 7T 1 H-MRS study. Neuropsychopharmacology 2012, 37, 2764–2771. [Google Scholar] [CrossRef] [Green Version]
- Linde, M.; Mulleners, W.M.; Chronicle, E.P.; McCrory, D.C. Gabapentin or pregabalin for the prophylaxis of episodic migraine in adults. Cochrane Database Syst. Rev. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez de la Aleja, J.; Ramos, A.; Mato-Abad, V.; Martínez-Salio, A.; Hernández-Tamames, J.A.; Molina, J.A.; Hernández-Gallego, J.; Álvarez-Linera, J. Higher glutamate to glutamine ratios in occipital regions in women with migraine during the interictal state. Headache J. Head Face Pain 2013, 53, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.M.; Pitchaimuthu, K.; Wu, Q.Z.; Carter, O.L.; Egan, G.F.; Badcock, D.R.; McKendrick, A.M. Relating excitatory and inhibitory neurochemicals to visual perception: A magnetic resonance study of occipital cortex between migraine events. PLoS ONE 2019, 14, e0208666. [Google Scholar] [CrossRef] [Green Version]
- Siniatchkin, M.; Sendacki, M.; Moeller, F.; Wolff, S.; Jansen, O.; Siebner, H.; Stephani, U. Abnormal changes of synaptic excitability in migraine with aura. Cereb. Cortex 2012, 22, 2207–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, J.; Sidoryk-Wegrzynowicz, M.; Zielinska, M.; Aschner, M. Roles of glutamine in neurotransmission. Neuron Glia Biol. 2010, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Kanai, R.; Chaieb, L.; Antal, A.; Walsh, V.; Paulus, W. Frequency-dependent electrical stimulation of the visual cortex. Curr. Biol. 2008, 18, 1839–1843. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.K.; Wade, A.R.; Penkman, K.E.; Baker, D.H. Dietary modulation of cortical excitation and inhibition. J. Psychopharmacol. 2017, 31, 632–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peroutka, S.J. What turns on a migraine? A systematic review of migraine precipitating factors. Curr. Pain Headache Rep. 2014, 18, 454. [Google Scholar] [CrossRef]
- Baldacci, F.; Vedovello, M.; Ulivi, M.; Vergallo, A.; Poletti, M.; Borelli, P.; Nuti, A.; Bonuccelli, U. How aware are migraineurs of their triggers? Headache J. Head Face Pain 2013, 53, 834–837. [Google Scholar] [CrossRef]
- Neut, D.; Fily, A.; Cuvellier, J.C.; Vallée, L. The prevalence of triggers in paediatric migraine: A questionnaire study in 102 children and adolescents. J. Headache Pain 2012, 13, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Spierings, E.L.; Ranke, A.H.; Honkoop, P.C. Precipitating and aggravating factors of migraine versus tension-type headache. Headache J. Head Face Pain 2001, 41, 554–558. [Google Scholar] [CrossRef]
- Shepherd, A.J. Visual stimuli, light and lighting are common triggers of migraine and headache. J. Light Vis. Environ. 2010, 34, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.J.; Chan, M.H.; Chen, L.; Wu, S.N.; Chen, H.H. Resveratrol attenuates cortical neuron activity: Roles of large conductance calcium-activated potassium channels and voltage-gated sodium channels. J. Biomed. Sci. 2016, 23, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peatfield, R.; Glover, V.; Littlewood, J.; Sandler, M.; Clifford Rose, F. The prevalence of diet-induced migraine. Cephalalgia 1984, 4, 179–183. [Google Scholar] [CrossRef]
- Nowaczewska, M.; Wiciński, M.; Kaźmierczak, W.; Kaźmierczak, H. To Eat or Not to eat: A Review of the Relationship between Chocolate and Migraines. Nutrients 2020, 12, 608. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Rodríguez, E.; Estrada-Zaldívar, B.; Zaldívar-Uribe, E. Effects of dark chocolate intake on brain electrical oscillations in healthy people. Foods 2018, 7, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marschollek, C.; Karimzadeh, F.; Jafarian, M.; Ahmadi, M.; Mohajeri, S.M.R.; Rahimi, S.; Speckmann, E.J.; Gorji, A. Effects of garlic extract on spreading depression: In vitro and investigations. Nutr. Neurosci. 2017, 20, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hougaard, A.; Amin, F.; Hauge, A.W.; Ashina, M.; Olesen, J. Provocation of migraine with aura using natural trigger factors. Neurology 2013, 80, 428–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowyer, S.M.; Aurora, S.K.; Moran, J.E.; Tepley, N.; Welch, K. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2001, 50, 582–587. [Google Scholar] [CrossRef]
- Al-Shimmery, E.K. Precipitating and relieving factors of migraine headache in 200 iraqi kurdish patients. Oman Med. J. 2010, 25, 212. [Google Scholar] [CrossRef]
- Martin, P.R. How do trigger factors acquire the capacity to precipitate headaches? Behav. Res. Ther. 2001, 39, 545–554. [Google Scholar] [CrossRef]
- Herrmann, C.S. Human EEG responses to 1–100 Hz flicker: Resonance phenomena in visual cortex and their potential correlation to cognitive phenomena. Exp. Brain Res. 2001, 137, 346–353. [Google Scholar] [CrossRef]
- Biglari, H.N.; Rezayi, A.; Biglari, H.N.; Alizadeh, M.; Ahmadabadi, F. Relationship between migraine and abnormal EEG findings in children. Iran. J. Child Neurol. 2012, 6, 21. [Google Scholar]
- Sand, T. Electroencephalography in migraine: A review with focus on quantitative electroencephalography and the migraine vs. epilepsy relationship. Cephalalgia 2003, 23, 5–11. [Google Scholar] [CrossRef]
- Drake Jr, M.E.; Bois, C.D.; Huber, S.J.; Pakalnis, A.; Denio, L.S. EEG spectral analysis and time domain descriptors in headache. Headache J. Head Face Pain 1988, 28, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Polich, J.; Ehlers, C.L.; Dalessio, D.J. Pattern-shift visual evoked responses and EEG in migraine. Headache J. Head Face Pain 1986, 26, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, V. Computerized EEG Topography in Childhood Headache. Cephalalgia 1987, 9. [Google Scholar] [CrossRef]
- Nyrke, T.; Kangasniemi, P.; Lang, H. Alpha rhythm in classical migraine (migraine with aura): Abnormalities in the headache-free interval. Cephalalgia 1990, 10, 177–181. [Google Scholar] [CrossRef]
- Jonkman, E.; Lelieveld, M. EEG computer analysis in patients with migraine. Electroencephalogr. Clin. Neurophysiol. 1981, 52, 652–655. [Google Scholar] [CrossRef]
- Facchetti, D.; Marsile, C.; Faggi, L.; Donati, E.; Kokodoko, A.; Poloni, M. Cerebral mapping in subjects suffering from migraine with aura. Cephalalgia 1990, 10, 279–284. [Google Scholar] [CrossRef]
- Bjørk, M.H.; Stovner, L.J.; Engstrøm, M.; Stjern, M.; Hagen, K.; Sand, T. Interictal quantitative EEG in migraine: A blinded controlled study. J. Headache Pain 2009, 10, 331. [Google Scholar] [CrossRef] [Green Version]
- Sand, T.; Zhitniy, N.; White, L.R.; Stovner, L.J. Visual evoked potential latency, amplitude and habituation in migraine: A longitudinal study. Clin. Neurophysiol. 2008, 119, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Sand, T.; White, L.; Hagen, K.; Stovner, L. Visual evoked potential and spatial frequency in migraine: A longitudinal study. Acta Neurol. Scand. 2009, 120, 33–37. [Google Scholar] [CrossRef]
- Shibata, K.; Osawa, M.; Iwata, M. Pattern reversal visual evoked potentials in migraine with aura and migraine aura without headache. Cephalalgia 1998, 18, 319–323. [Google Scholar] [CrossRef]
- Evers, S.; Quibeldey, F.; Grotemeyer, K.H.; Suhr, B.; Husstedt, I. Dynamic changes of cognitive habituation and serotonin metabolism during the migraine interval. Cephalalgia 1999, 19, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Judit, A.; Sandor, P.; Schoenen, J. Habituation of visual and intensity dependence of auditory evoked cortical potentials tends to normalize just before and during the migraine attack. Cephalalgia 2000, 20, 714–719. [Google Scholar] [CrossRef]
- Shahaf, G.; Kuperman, P.; Bloch, Y.; Yariv, S.; Granovsky, Y. Monitoring migraine cycle dynamics with an easy-to-use electrophysiological marker—A pilot study. Sensors 2018, 18, 3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dow, D.J.; Whitty, C. Electro-encephalographic changes in migraine review of 51 cases. Lancet 1947, 250, 52–54. [Google Scholar] [CrossRef]
- Malik, A.B.; Alptekin, B.; Bajwa, Z.H. Chapter 34—Classification of Headache. In Essentials of Pain Medicine and Regional Anesthesia, 2nd ed.; Benzon, H.T., Raja, S.N., Molloy, R.E., Liu, S.S., Fishman, S.M., Eds.; Churchill Livingstone: Philadelphia, PA, USA, 2005; pp. 277–281. [Google Scholar] [CrossRef]
- Ganji, S. Basilar artery migraine: EEG and evoked potential patterns during acute stage. Headache J. Head Face Pain 1986, 26, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Spina, I.L.; Vignati, A.; Porazzi, D. Basilar artery migraine: Transcranial Doppler EEG and SPECT from the aura phase to the end. Headache J. Head Face Pain 1997, 37, 43–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriani, S.; Scarpa, P.; Faggioli, R.; Carlo, L.D.; Voghenzi, A. Uncommon EEG Pattern in an 8-Year-Old Boy With Recurrent Migraine Aura Without Headache. Headache J. Head Face Pain 1993, 33, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Pisani, F.; Fusco, C. Ictal and interictal EEG findings in children with migraine. J. Headache Pain 2004, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Ramelli, G.; Sturzenegger, M.; Donati, F.; Karbowski, K. EEG findings during basilar migraine attacks in children. Electroencephalogr. Clin. Neurophysiol. 1998, 107, 374–378. [Google Scholar] [CrossRef]
- Sansone, M.; Marinelli, G.; Piccotti, E.; Severino, M.; Nobili, L. Cerebral blood flow in a case of typical aura without headache. J. Neurol. 2019, 266, 2869–2871. [Google Scholar] [CrossRef]
- Tan, H.; Suganthi, C.; Dhachayani, S.; Rizal, A.; Raymond, A. The electroencephalogram changes in migraineurs. Med. J. Malays. 2007, 62, 56–58. [Google Scholar]
- Woods, R.P.; Iacoboni, M.; Mazziotta, J.C. Bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. N. Engl. J. Med. 1994, 331, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M.; Olsen, T.S.; Lassen, N.A.; Paulson, O.B. Regulation of regional cerebral blood flow during and between migraine attacks. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1983, 14, 569–572. [Google Scholar] [CrossRef]
- Friberg, L.; Olesen, J.; Lassen, N.A.; Olsen, T.S.; Karle, A. Cerebral oxygen extraction, oxygen consumption, and regional cerebral blood flow during the aura phase of migraine. Stroke 1994, 25, 974–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arngrim, N.; Schytz, H.W.; Britze, J.; Amin, F.M.; Vestergaard, M.B.; Hougaard, A.; Wolfram, F.; de Koning, P.J.; Olsen, K.S.; Secher, N.H.; et al. Migraine induced by hypoxia: An MRI spectroscopy and angiography study. Brain 2016, 139, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Blank Jr, W.; Kirshner, H. The kinetics of extracellular potassium changes during hypoxia and anoxia in the cat cerebral cortex. Brain Res. 1977, 123, 113–124. [Google Scholar] [CrossRef]
- Lee, C.H.; Seo, M.W.; Shin, B.S.; Yang, T.H.; Shin, H.J.; Ryu, H.U. Intermittent theta slowings in contralateral side of weakness after sleep deprivation on spot EEG in sporadic hemiplegic migraine. J. Epilepsy Res. 2016, 6, 102. [Google Scholar] [CrossRef] [Green Version]
- Rey, V.; Aybek, S.; Maeder-Ingvar, M.; Rossetti, A.O. Positive occipital sharp transients of sleep (POSTS): A reappraisal. Clin. Neurophysiol. 2009, 120, 472–475. [Google Scholar] [CrossRef]
- Speckman, E.; Elger, C.; Gorji, A. Neurophysiologic Basis of EEG and DC Potentials. In Neidermeyer’s Electroencephalography; Schomer, D.L., Lopes da Silva, F.H., Eds.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Caspers, H.; Speckmann, E.J. DC Shifts, EEG Waves and Neuronal Membrane Potentials in the Cat Cerebral Cortex during Seizure Activity. In The Responsive Brain; Elsevier: Amsterdam, The Netherlands, 1976; pp. 195–199. [Google Scholar]
- Áfra, J.; Mascia, A.; Phy, P.G.; De Noordhout, A.M.; Schoenen, J. Interictal cortical excitability in migraine: A study using transcranial magnetic stimulation of motor and visual cortices. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1998, 44, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Brighina, F.; Piazza, A.; Daniele, O.; Fierro, B. Modulation of visual cortical excitability in migraine with aura: Effects of 1 Hz repetitive transcranial magnetic stimulation. Exp. Brain Res. 2002, 145, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Akben, S.B.; Subasi, A.; Tuncel, D. Analysis of EEG signals under flash stimulation for migraine and epileptic patients. J. Med. Syst. 2011, 35, 437–443. [Google Scholar] [CrossRef]
- Schoenen, J.; Jamart, B.; Delwaide, P. Cartographie electroencephalographique dans les migraines en periodes critique et intercritique. Rev. d’éLectroencéphalographie Neurophysiol. Clin. 1987, 17, 289–299. [Google Scholar] [CrossRef]
- Hall, S.D.; Barnes, G.R.; Hillebrand, A.; Furlong, P.L.; Singh, K.D.; Holliday, I.E. Spatio-temporal imaging of cortical desynchronization in migraine visual aura: A magnetoencephalography case study. Headache J. Head Face Pain 2004, 44, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Seri, S.; Cerquiglini, A.; Guidetti, V. Computerized EEG topography in childhood migraine between and during attacks. Cephalalgia 1993, 13, 53–56. [Google Scholar] [CrossRef]
- daSilva Morgan, K.; Elder, G.J.; Collerton, D.; Taylor, J.P. The utility and application of electrophysiological methods in the study of visual hallucinations. Clin. Neurophysiol. 2018, 129, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Klimesch, W.; Sauseng, P.; Hanslmayr, S. EEG alpha oscillations: The inhibition-timing hypothesis. Brain Res. Rev. 2007, 53, 63–88. [Google Scholar] [CrossRef] [PubMed]
- Dugué, L.; Marque, P.; VanRullen, R. The phase of ongoing oscillations mediates the causal relation between brain excitation and visual perception. J. Neurosci. 2011, 31, 11889–11893. [Google Scholar] [CrossRef] [PubMed]
- Jensen, O.; Gips, B.; Bergmann, T.O.; Bonnefond, M. Temporal coding organized by coupled alpha and gamma oscillations prioritize visual processing. Trends Neurosci. 2014, 37, 357–369. [Google Scholar] [CrossRef]
- Lorincz, M.L.; Kékesi, K.A.; Juhász, G.; Crunelli, V.; Hughes, S.W. Temporal framing of thalamic relay-mode firing by phasic inhibition during the alpha rhythm. Neuron 2009, 63, 683–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgarten, T.J.; Neugebauer, J.; Oeltzschner, G.; Füllenbach, N.D.; Kircheis, G.; Häussinger, D.; Lange, J.; Wittsack, H.J.; Butz, M.; Schnitzler, A. Connecting occipital alpha band peak frequency, visual temporal resolution, and occipital GABA levels in healthy participants and hepatic encephalopathy patients. NeuroImage Clin. 2018, 20, 347–356. [Google Scholar] [CrossRef]
- Shevelev, I.; Kamenkovich, V.; Bark, E.; Verkhlutov, V.; Sharaev, G.; Mikhailova, E. Visual illusions and travelling alpha waves produced by flicker at alpha frequency. Int. J. Psychophysiol. 2000, 39, 9–20. [Google Scholar] [CrossRef]
- Walter, W.G. The Living Brain; WW Norton: New York, NY, USA, 1953. [Google Scholar]
- Walter, W.G. Colour illusions and aberrations during stimulation by flickering light. Nature 1956, 177, 710. [Google Scholar] [CrossRef] [PubMed]
- Walter, V.; Walter, W. The central effects of rhythmic sensory stimulation. Electroencephalogr. Clin. Neurophysiol. 1949, 1, 57–86. [Google Scholar] [CrossRef]
- Crotogino, J.; Feindel, A.; Wilkinson, F. Perceived scintillation rate of migraine aura. Headache J. Head Face Pain 2001, 41, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Mauro, F.; Raffone, A.; VanRullen, R. A bidirectional link between brain oscillations and geometric patterns. J. Neurosci. 2015, 35, 7921–7926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, J.; Chiou, R.; Rogers, S.; Wicken, M.; Heitmann, S.; Ermentrout, B. Sensory dynamics of visual hallucinations in the normal population. Elife 2016, 5, e17072. [Google Scholar] [CrossRef] [PubMed]
- Gert van Dijk, J.; Haan, J.; Ferrari, M. Photic stimulation and the diagnosis of migraine. Headache Q. 1992, 3, 387. [Google Scholar]
- Fogang, Y.; Gérard, P.; De Pasqua, V.; Pepin, J.L.; Ndiaye, M.; Magis, D.; Schoenen, J. Analysis and clinical correlates of 20 Hz photic driving on routine EEG in migraine. Acta Neurol. Belg. 2015, 115, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Shibata, K.; Yamane, K.; Otuka, K.; Iwata, M. Abnormal visual processing in migraine with aura: A study of steady-state visual evoked potentials. J. Neurol. Sci. 2008, 271, 119–126. [Google Scholar] [CrossRef]
- Shibata, K.; Yamane, K.; Nishimura, Y.; Kondo, H.; Otuka, K. Spatial frequency differentially affects habituation in migraineurs: A steady-state visual-evoked potential study. Doc. Ophthalmol. 2011, 123, 65. [Google Scholar] [CrossRef]
- Shiina, T.; Takashima, R.; Pascual-Marqui, R.D.; Suzuki, K.; Watanabe, Y.; Hirata, K. Evaluation of electroencephalogram using exact low-resolution electromagnetic tomography during photic driving response in patients with migraine. Neuropsychobiology 2019, 77, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Graaf, T.A.; Gross, J.; Paterson, G.; Rusch, T.; Sack, A.T.; Thut, G. Alpha-band rhythms in visual task performance: Phase-locking by rhythmic sensory stimulation. PLoS ONE 2013, 8, e60035. [Google Scholar] [CrossRef]
- Keitel, C.; Quigley, C.; Ruhnau, P. Stimulus-driven brain oscillations in the alpha range: Entrainment of intrinsic rhythms or frequency-following response? J. Neurosci. 2014, 34, 10137–10140. [Google Scholar] [CrossRef] [Green Version]
- Gerster, M.; Berner, R.; Sawicki, J.; Zakharova, A.; Škoch, A.; Hlinka, J.; Lehnertz, K.; Schöll, E. FitzHugh–Nagumo oscillators on complex networks mimic epileptic-seizure-related synchronization phenomena. Chaos Interdiscip. J. Nonlinear Sci. 2020, 30, 123130. [Google Scholar]
- De Tommaso, M.; Stramaglia, S.; Marinazzo, D.; Trotta, G.; Pellicoro, M. Functional and effective connectivity in EEG alpha and beta bands during intermittent flash stimulation in migraine with and without aura. Cephalalgia 2013, 33, 938–947. [Google Scholar] [CrossRef]
- Trotta, G.; Stramaglia, S.; Pellicoro, M.; Bellotti, R.; Marinazzo, D.; de Tommaso, M. Effective connectivity and cortical information flow under visual stimulation in migraine with aura. In Proceedings of the 5th IEEE International Workshop on Advances in Sensors and Interfaces IWASI, Bari, Italy, 13–14 June 2013; pp. 228–232. [Google Scholar]
- de Tommaso, M.; Trotta, G.; Vecchio, E.; Ricci, K.; Siugzdaite, R.; Stramaglia, S. Brain networking analysis in migraine with and without aura. J. Headache Pain 2017, 18, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Tommaso, M.; Trotta, G.; Vecchio, E.; Ricci, K.; Van de Steen, F.; Montemurno, A.; Lorenzo, M.; Marinazzo, D.; Bellotti, R.; Stramaglia, S. Functional connectivity of EEG signals under laser stimulation in migraine. Front. Hum. Neurosci. 2015, 9, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Y.; Zheng, R.; Li, K.; Zhang, Y.; Lyu, D.; Jia, H.; Ren, Y.; Zou, Y. The altered Granger causality connection among pain-related brain networks in migraine. Medicine 2018, 97, e0102. [Google Scholar] [CrossRef]
- Martins, I.P.; Westerfield, M.; Lopes, M.; Maruta, C.; Gil-da Costa, R. Brain state monitoring for the future prediction of migraine attacks. Cephalalgia 2020, 40, 255–265. [Google Scholar] [CrossRef]
- O’Hare, L.; Hibbard, P.B. Visual processing in migraine. Cephalalgia 2016, 36, 1057–1076. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, A.J. Increased visual after-effects following pattern adaptation in migraine: A lack of intracortical excitation? Brain 2001, 124, 2310–2318. [Google Scholar] [CrossRef]
- Wagner, D.; Manahilov, V.; Loffler, G.; Gordon, G.E.; Dutton, G.N. Visual noise selectively degrades vision in migraine. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2294–2299. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.; Manahilov, V.; Gordon, G.E.; Loffler, G. Global shape processing deficits are amplified by temporal masking in migraine. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1160–1168. [Google Scholar] [CrossRef] [Green Version]
- Hammett, S.T.; Cook, E.; Hassan, O.; Hughes, C.A.; Rooslien, H.; Tizkar, R.; Larsson, J. GABA, noise and gain in human visual cortex. Neurosci. Lett. 2020, 736, 135294. [Google Scholar] [CrossRef]
- Shepherd, A. Visual contrast processing in migraine. Cephalalgia 2000, 20, 865–880. [Google Scholar] [CrossRef]
- Asher, J.M.; O’Hare, L.; Romei, V.; Hibbard, P.B. Typical lateral interactions, but increased contrast sensitivity, in migraine-with-aura. Vision 2018, 2, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldrich, A.; Hibbard, P.; Wilkins, A. Vision and hyper-responsiveness in migraine. Vision 2019, 3, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKendrick, A.; Sampson, G. Low spatial frequency contrast sensitivity deficits in migraine are not visual pathway selective. Cephalalgia 2009, 29, 539–549. [Google Scholar] [CrossRef]
- Yoon, J.H.; Maddock, R.J.; Rokem, A.; Silver, M.A.; Minzenberg, M.J.; Ragland, J.D.; Carter, C.S. GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression. J. Neurosci. 2010, 30, 3777–3781. [Google Scholar] [CrossRef] [Green Version]
- Edden, R.A.; Muthukumaraswamy, S.D.; Freeman, T.C.; Singh, K.D. Orientation discrimination performance is predicted by GABA concentration and gamma oscillation frequency in human primary visual cortex. J. Neurosci. 2009, 29, 15721–15726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKendrick, A.M.; Vingrys, A.J.; Badcock, D.R.; Heywood, J.T. Visual dysfunction between migraine events. Investig. Ophthalmol. Vis. Sci. 2001, 42, 626–633. [Google Scholar]
- Tibber, M.S.; Guedes, A.; Shepherd, A.J. Orientation discrimination and contrast detection thresholds in migraine for cardinal and oblique angles. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5599–5604. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, F.; Crotogino, J. Orientation discrimination thresholds in migraine: A measure of visual cortical inhibition. Cephalalgia 2000, 20, 57–66. [Google Scholar] [CrossRef]
- Pitchaimuthu, K.; Wu, Q.Z.; Carter, O.; Nguyen, B.N.; Ahn, S.; Egan, G.F.; McKendrick, A.M. Occipital GABA levels in older adults and their relationship to visual perceptual suppression. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battista, J.; Badcock, D.R.; McKendrick, A.M. Migraine increases centre-surround suppression for drifting visual stimuli. PLoS ONE 2011, 6, e18211. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, P.; Serrano-Pedraza, I.; Whittaker, R.G.; Trevelyan, A.; Read, J.C. Two common psychophysical measures of surround suppression reflect independent neuronal mechanisms. J. Vis. 2015, 15, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schallmo, M.P.; Millin, R.; Kale, A.M.; Kolodny, T.; Edden, R.A.; Bernier, R.A.; Murray, S.O. Glutamatergic facilitation of neural responses in MT enhances motion perception in humans. NeuroImage 2019, 184, 925–931. [Google Scholar] [CrossRef]
- Takeuchi, T.; Yoshimoto, S.; Shimada, Y.; Kochiyama, T.; Kondo, H.M. Individual differences in visual motion perception and neurotransmitter concentrations in the human brain. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160111. [Google Scholar] [CrossRef] [Green Version]
- Antal, A.; Temme, J.; Nitsche, M.; Varga, E.; Lang, N.; Paulus, W. Altered motion perception in migraineurs: Evidence for interictal cortical hyperexcitability. Cephalalgia 2005, 25, 788–794. [Google Scholar] [CrossRef]
- Tibber, M.S.; Kelly, M.G.; Jansari, A.; Dakin, S.C.; Shepherd, A.J. An inability to exclude visual noise in migraine. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2539–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, A.J.; Wyatt, G.; Tibber, M.S. Visual metacontrast masking in migraine. Cephalalgia 2011, 31, 346–356. [Google Scholar] [CrossRef]
- Drummond, P.D.; Anderson, M. Visual field loss after attacks of migraine with aura. Cephalalgia 1992, 12, 349–352. [Google Scholar] [CrossRef] [Green Version]
- McKendrick, A.M.; Badcock, D.R. Decreased visual field sensitivity measured 1 day, then 1 week, after migraine. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1061–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, B.N.; Vingrys, A.J.; McKendrick, A.M. The effect of duration post-migraine on visual electrophysiology and visual field performance in people with migraine. Cephalalgia 2014, 34, 42–57. [Google Scholar] [CrossRef] [PubMed]
- McKendrick, A.M.; Chan, Y.M.; Vingrys, A.J.; Turpin, A.; Badcock, D.R. Daily vision testing can expose the prodromal phase of migraine. Cephalalgia 2018, 38, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Luedtke, K.; Schulte, L.H.; May, A. Visual processing in migraineurs depends on the migraine cycle. Ann. Neurol. 2019, 85, 280–283. [Google Scholar] [CrossRef]
- Schulte, L.H.; May, A. The migraine generator revisited: Continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain 2016, 139, 1987–1993. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, A.J. Tracking the migraine cycle using visual tasks. Vision 2020, 4, 23. [Google Scholar] [CrossRef]
- Ergenoglu, T.; Demiralp, T.; Bayraktaroglu, Z.; Ergen, M.; Beydagi, H.; Uresin, Y. Alpha rhythm of the EEG modulates visual detection performance in humans. Cogn. Brain Res. 2004, 20, 376–383. [Google Scholar] [CrossRef]
- Hanslmayr, S.; Klimesch, W.; Sauseng, P.; Gruber, W.; Doppelmayr, M.; Freunberger, R.; Pecherstorfer, T. Visual discrimination performance is related to decreased alpha amplitude but increased phase locking. Neurosci. Lett. 2005, 375, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Hanslmayr, S.; Aslan, A.; Staudigl, T.; Klimesch, W.; Herrmann, C.S.; Bäuml, K.H. Prestimulus oscillations predict visual perception performance between and within subjects. Neuroimage 2007, 37, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Osawa, M.; Iwata, M. Pattern reversal visual evoked potentials in classic and common migraine. J. Neurol. Sci. 1997, 145, 177–181. [Google Scholar] [CrossRef]
- Shibata, K.; Yamane, K.; Iwata, M.; Ohkawa, S. Evaluating the effects of spatial frequency on migraines by using pattern-reversal visual evoked potentials. Clin. Neurophysiol. 2005, 116, 2220–2227. [Google Scholar] [CrossRef]
- O’Hare, L.; Menchinelli, F.; Durrant, S.J. Resting-state alpha-band oscillations in migraine. Perception 2018, 47, 379–396. [Google Scholar] [CrossRef]
- Cao, Z. Developing a Migraine Attack Prediction System Using Resting-State EEG. Ph.D. Thesis, University of Technology, Sydney, Australia, 2017. [Google Scholar]
- Borgdorff, P. Arguments against the role of cortical spreading depression in migraine. Neurol. Res. 2018, 40, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Bolay, H.; Vuralli, D.; Goadsby, P.J. Aura and Head pain: Relationship and gaps in the translational models. J. Headache Pain 2019, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef]
- Kroos, J.M.; Marinelli, I.; Diez, I.; Cortes, J.M.; Stramaglia, S.; Gerardo-Giorda, L. Patient-specific computational modeling of cortical spreading depression via diffusion tensor imaging. Int. J. Numer. Methods Biomed. Eng. 2017, 33, e2874. [Google Scholar] [CrossRef] [Green Version]
- Kroos, J.M.; de Tommaso, M.; Stramaglia, S.; Vecchio, E.; Burdi, N.; Gerardo-Giorda, L. Clinical correlates of mathematical modeling of cortical spreading depression: Single-cases study. Brain Behav. 2019, 9, e01387. [Google Scholar] [CrossRef]
- Borsook, D.; Maleki, N.; Becerra, L.; McEwen, B. Understanding migraine through the lens of maladaptive stress responses: A model disease of allostatic load. Neuron 2012, 73, 219–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigal, M.E.; Lipton, R.B. Concepts and mechanisms of migraine chronification. Headache J. Head Face Pain 2008, 48, 7–15. [Google Scholar] [CrossRef]
- Kunkel, R.S. Migraine aura without headache: Benign, but a diagnosis of exclusion. Clevel. Clin. J. Med. 2005, 72, 529. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J. International classification of headache disorders. Lancet Neurol. 2018, 17, 396–397. [Google Scholar] [CrossRef] [Green Version]
- Chronicle, E.; Mulleners, W. Might migraine damage the brain? Cephalalgia 1994, 14, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.M.; Legg, N.J.; Anderson, D.J. Long term decline of P100 amplitude in migraine with aura. J. Neurol. Neurosurg. Psychiatry 2000, 69, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Rocca, M.A.; Ceccarelli, A.; Falini, A.; Colombo, B.; Tortorella, P.; Bernasconi, L.; Comi, G.; Scotti, G.; Filippi, M. Brain gray matter changes in migraine patients with T2-visible lesions: A 3-T MRI study. Stroke 2006, 37, 1765–1770. [Google Scholar] [CrossRef] [Green Version]
- Kurth, T.; Diener, H.C. Current views of the risk of stroke for migraine with and migraine without aura. Curr. Pain Headache Rep. 2006, 10, 214–220. [Google Scholar] [CrossRef]
- Hartings, J.A.; Shuttleworth, C.W.; Kirov, S.A.; Ayata, C.; Hinzman, J.M.; Foreman, B.; Andrew, R.D.; Boutelle, M.G.; Brennan, K.; Carlson, A.P.; et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leao’s legacy. J. Cereb. Blood Flow Metab. 2017, 37, 1571–1594. [Google Scholar] [CrossRef] [Green Version]
- Orr, S.L.; Kabbouche, M.A.; O’Brien, H.L.; Kacperski, J.; Powers, S.W.; Hershey, A.D. Paediatric migraine: Evidence-based management and future directions. Nat. Rev. Neurol. 2018, 14, 515–527. [Google Scholar] [CrossRef]
- Merikangas, K.R. Contributions of epidemiology to our understanding of migraine. Headache J. Head Face Pain 2013, 53, 230–246. [Google Scholar] [CrossRef]
- Balestri, M.; Papetti, L.; Maiorani, D.; Capuano, A.; Tarantino, S.; Battan, B.; Vigevano, F.; Valeriani, M. Features of aura in paediatric migraine diagnosed using the ICHD 3 beta criteria. Cephalalgia 2018, 38, 1742–1747. [Google Scholar] [CrossRef] [PubMed]
- Victor, T.; Hu, X.; Campbell, J.; Buse, D.; Lipton, R. Migraine prevalence by age and sex in the United States: A life-span study. Cephalalgia 2010, 30, 1065–1072. [Google Scholar] [CrossRef]
- Stewart, W.; Wood, C.; Reed, M.; Roy, J.; Lipton, R. Cumulative lifetime migraine incidence in women and men. Cephalalgia 2008, 28, 1170–1178. [Google Scholar] [CrossRef]
- MacGregor, E.A. Migraine, menopause and hormone replacement therapy. Post Reprod. Health 2018, 24, 11–18. [Google Scholar] [CrossRef]
- Stewart, W.F.; Lipton, R.; Chee, E.; Sawyer, J.; Silberstein, S. Menstrual cycle and headache in a population sample of migraineurs. Neurology 2000, 55, 1517–1523. [Google Scholar] [CrossRef]
- Ripa, P.; Ornello, R.; Degan, D.; Tiseo, C.; Stewart, J.; Pistoia, F.; Carolei, A.; Sacco, S. Migraine in menopausal women: A systematic review. Int. J. Women’s Health 2015, 7, 773. [Google Scholar]
- MacGregor, E.A. Oestrogen and attacks of migraine with and without aura. Lancet Neurol. 2004, 3, 354–361. [Google Scholar] [CrossRef]
- Mattsson, P. Hormonal factors in migraine: A population-based study of women aged 40 to 74 years. Headache J. Head Face Pain 2003, 43, 27–35. [Google Scholar] [CrossRef]
- Kopell, B.S.; Lunde, D.T.; Clayton, R.B.; Moos, R.H. Variations in some measures of arousal during the menstrual cycle. J. Nerv. Ment. Dis. 1969, 148, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Parlee, M.B. Menstrual rhythm in sensory processes: A review of fluctuations in vision, olfaction, audition, taste, and touch. Psychol. Bull. 1983, 93, 539. [Google Scholar] [CrossRef] [PubMed]
- DeMarchi, G.; Tong, J. Menstrual, diurnal, and activation effects on the resolution of temporally paired flashes. Psychophysiology 1972, 9, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.; Diamond, A.L.; Mast, M. Visual sensitivity and sexual arousal levels during the menstrual cycle. J. Nerv. Ment. Dis. 1972, 155, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.M.; Stone, S.C.; Sandman, C.A. Visual perception in women during the menstrual cycle. Physiol. Behav. 1978, 20, 239–243. [Google Scholar] [CrossRef]
- Barris, M.; Dawson, W.; Theiss, C. The visual sensitivity of women during the menstrual cycle. Doc. Ophthalmol. 1980, 49, 293–301. [Google Scholar] [CrossRef]
- Scher, D.; Pionk, M.; Purcell, D.G. Visual sensitivity fluctuations during the menstrual cycle under dark and light adaptation. Bull. Psychon. Soc. 1981, 18, 159–160. [Google Scholar] [CrossRef]
- Johnson, N.; Petersik, J.T. Preliminary findings suggesting cyclic changes in visual contrast sensitivity during the menstrual cycle. Percept. Mot. Skills 1987, 64, 587–594. [Google Scholar] [CrossRef]
- Webb, A.L.; Hibbard, P.B.; O’Gorman, R. Natural variation in female reproductive hormones does not affect contrast sensitivity. R. Soc. Open Sci. 2018, 5, 171566. [Google Scholar] [CrossRef] [Green Version]
- Deza, L.; Eidelberg, E. Development of cortical electrical activity in the rat. Exp. Neurol. 1967, 17, 425–438. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Hare, L.; Asher, J.M.; Hibbard, P.B. Migraine Visual Aura and Cortical Spreading Depression—Linking Mathematical Models to Empirical Evidence. Vision 2021, 5, 30. https://doi.org/10.3390/vision5020030
O’Hare L, Asher JM, Hibbard PB. Migraine Visual Aura and Cortical Spreading Depression—Linking Mathematical Models to Empirical Evidence. Vision. 2021; 5(2):30. https://doi.org/10.3390/vision5020030
Chicago/Turabian StyleO’Hare, Louise, Jordi M. Asher, and Paul B. Hibbard. 2021. "Migraine Visual Aura and Cortical Spreading Depression—Linking Mathematical Models to Empirical Evidence" Vision 5, no. 2: 30. https://doi.org/10.3390/vision5020030
APA StyleO’Hare, L., Asher, J. M., & Hibbard, P. B. (2021). Migraine Visual Aura and Cortical Spreading Depression—Linking Mathematical Models to Empirical Evidence. Vision, 5(2), 30. https://doi.org/10.3390/vision5020030