1. Introduction
Dengue viruses (DV) are mosquito-borne single-stranded RNA viruses within the family
Flaviviridae. DV infections have become one of the most common diseases causing morbidity and mortality affecting millions of individuals worldwide [
1]. The viruses, upon entering the cells, shed their coats and release their positive stranded RNA into the cytoplasm and begin translation into proteins. The process induces unique changes of the endoplasmic reticulum (ER) facilitating the replication of the RNA and packaging and exporting new virions. Recent applications of advanced electron microscopic technologies in the in vitro studies of flavivirus-infected cells have defined a set of distinct virus-induced endoplasmic membrane structures specific for DV and closely associated viruses including West Nile and Tick Borne Encephalitis [
2]. The advanced studies have also described the functional aspects and interactions between the viruses and host molecular systems (reviewed in [
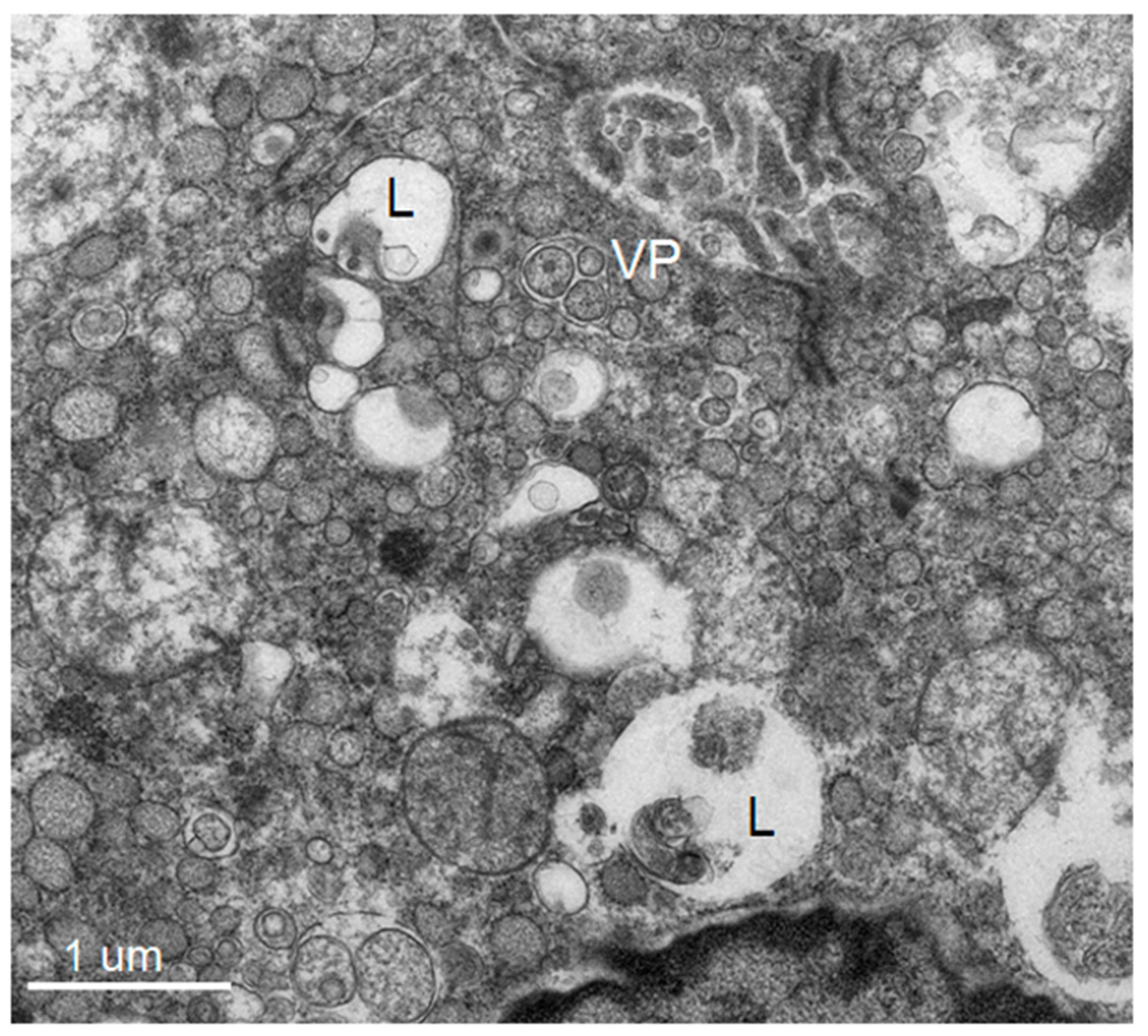
3]). The structural features include: membrane invaginations into the ER lumen forming intra luminal vesicles (Ve) of ~90 nm which accumulate as vesicle packets (VP) (some vesicles maintain pore-like connections to the cytoplasm) and collections of convoluted membrane structures (CM) and membrane tubules (T) [
4,
5]. Virions are found within the ER membrane cisternae. These induced structures function as viral replicating factories where RNA translation, replication and packaging of the viral genome into virions take place [
3]. Each new structure provides a particular function: Ve serves as a site for the “viral replicating complex” and a protected environment against host innate immune reactions to newly synthesized double stranded RNA [
5,
6], while CM is a site for RNA translation and viral protein processing [
5,
7]. The pattern of these cytoplasmic changes is found to be unique amongst closely related viruses within the same genera infecting mammalian and insect cells in vitro [
5,
6]. However, ultrastructural studies have not been carried out using in vivo infection models, where cell functions are under the influence of host physiological mechanisms.
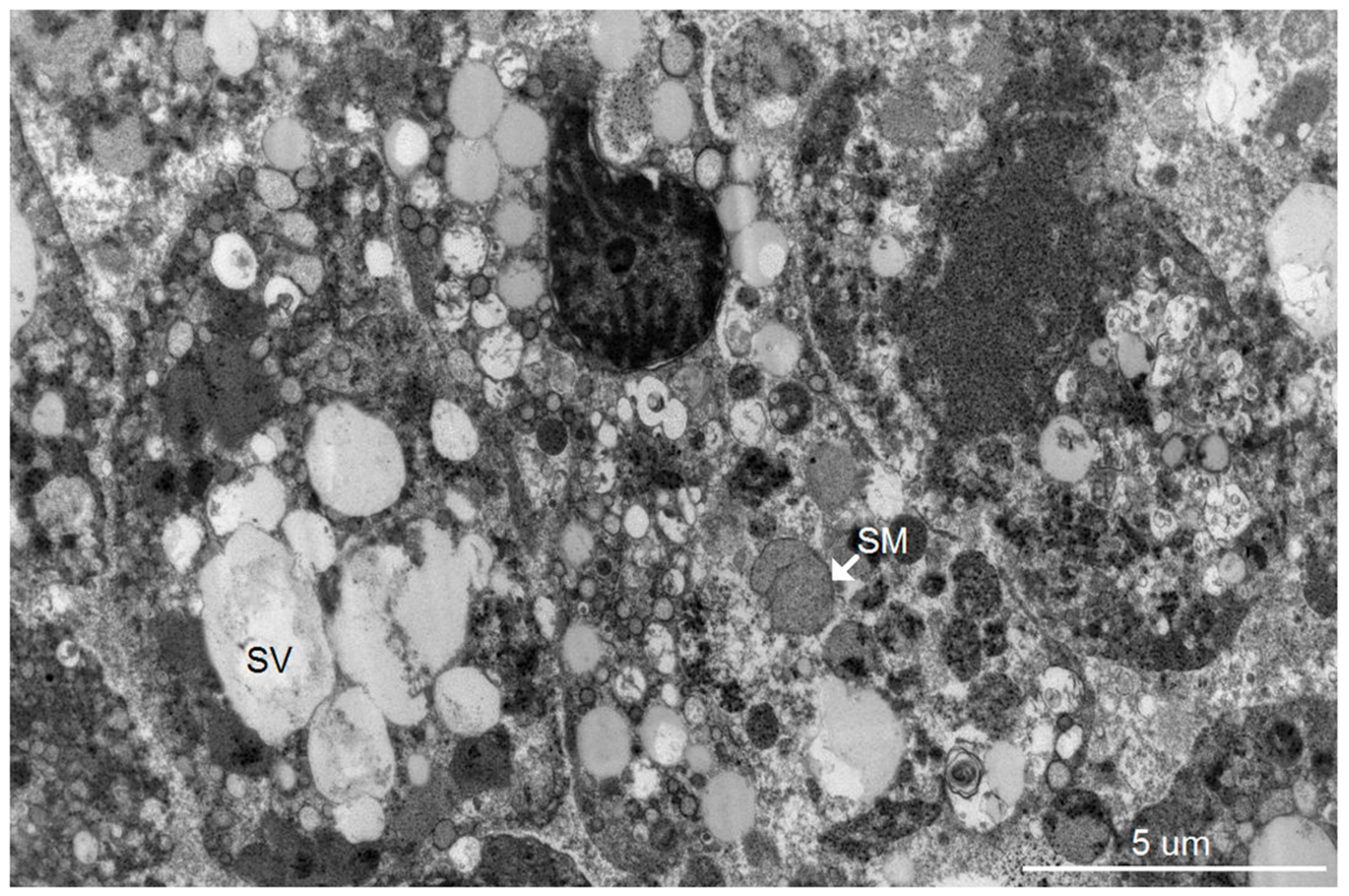
Our group has previously reported an autopsy study of 13 patients who died of severe dengue hemorrhagic fever (DHF) [
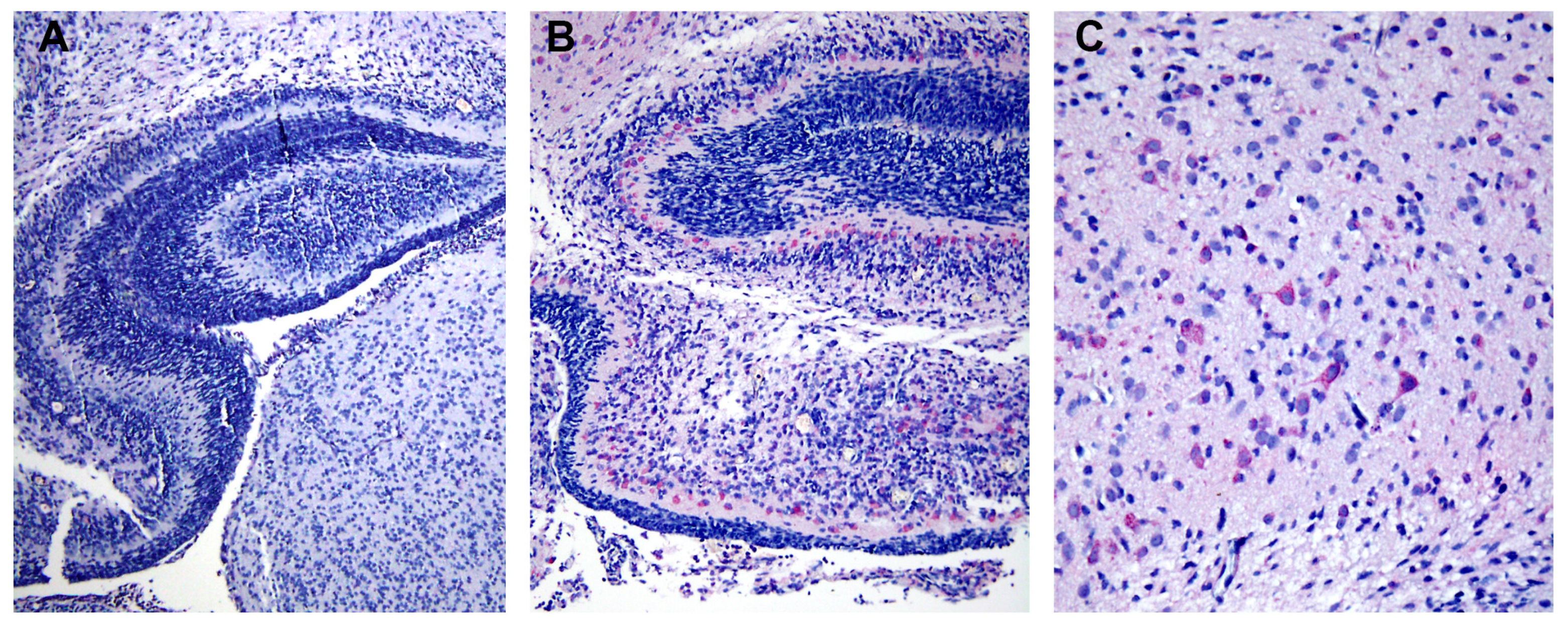
8]. Using light microscopic and immunohistochemistry techniques, we have identified three major organs, i.e., liver, spleen and lymph nodes, that had significant levels of DV- RNA and – antigens, as well as histopathological changes. To further investigate ultrastructural changes in the human liver tissues, the present study used the same set of samples for transmission electron microscopy and searched for histopathology and modification of ER membrane structures, which are indicative of active viral replication, in the autopsy specimens. In addition, electron microscopic (EM) studies of brain tissues from suckling mice intra-cerebrally infected with DV were carried out in parallel to define the in vivo cytoplasmic structural changes during acute DV infection. The results showed scant features of endoplasmic changes and no obvious evidence of virion production in hepatocytes from DHF patients, but instead displayed abundant steatotic hepatic cell damages. Suckling mouse brains (SMB) infected with DV, on the other hand, showed typical features of ER changes in neurons and microglia similar to the observation in the in vitro hepatocyte infection model [
2]. Some unique features of ER changes were also identified.
4. Discussion
Our study is the most comprehensive ultrastructural study of the liver from patients who died of severe DHF. Specimens designed for EM study from seven patients were studied. The study was a follow up from the histopathology study reported in 2014 by Aye et al., using materials systematically and properly collected from a group of children who died of DHF/DSS [
8]. In the previous study, the liver was one of the key organs found to be infected. DV antigens (both structural and non-structural) were identified in hepatocytes by immunohistochemistry methods; DV-specific RNAs were also detected and quantified from liver tissue extracts. The degree of damage to the liver has been systematically studied and quantified. Two surprising findings in this current study are the fact that there were very few cytoplasmic structural changes compatible with efficient virus reproduction as those found in the suckling mouse brain, despite overwhelming evidence of the presence of viral antigens and RNAs in the tissues at the time of autopsy [
8]. In addition, the pattern of responses by patients’ liver cells is completely different from the manner in which the hepatocyte cell lines, i.e., HepG2 or Huh-7 responded in vitro to the viruses [
5].
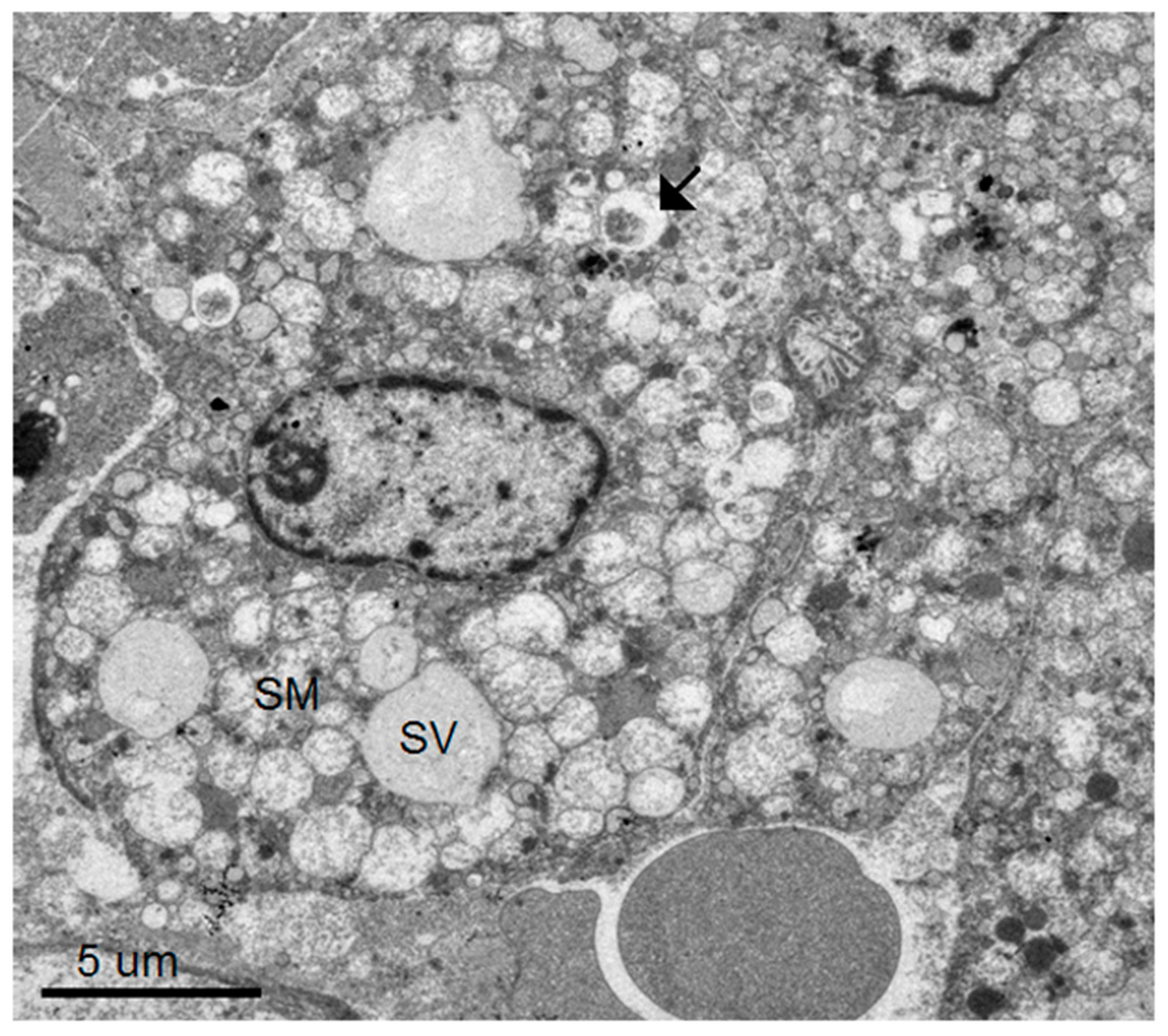
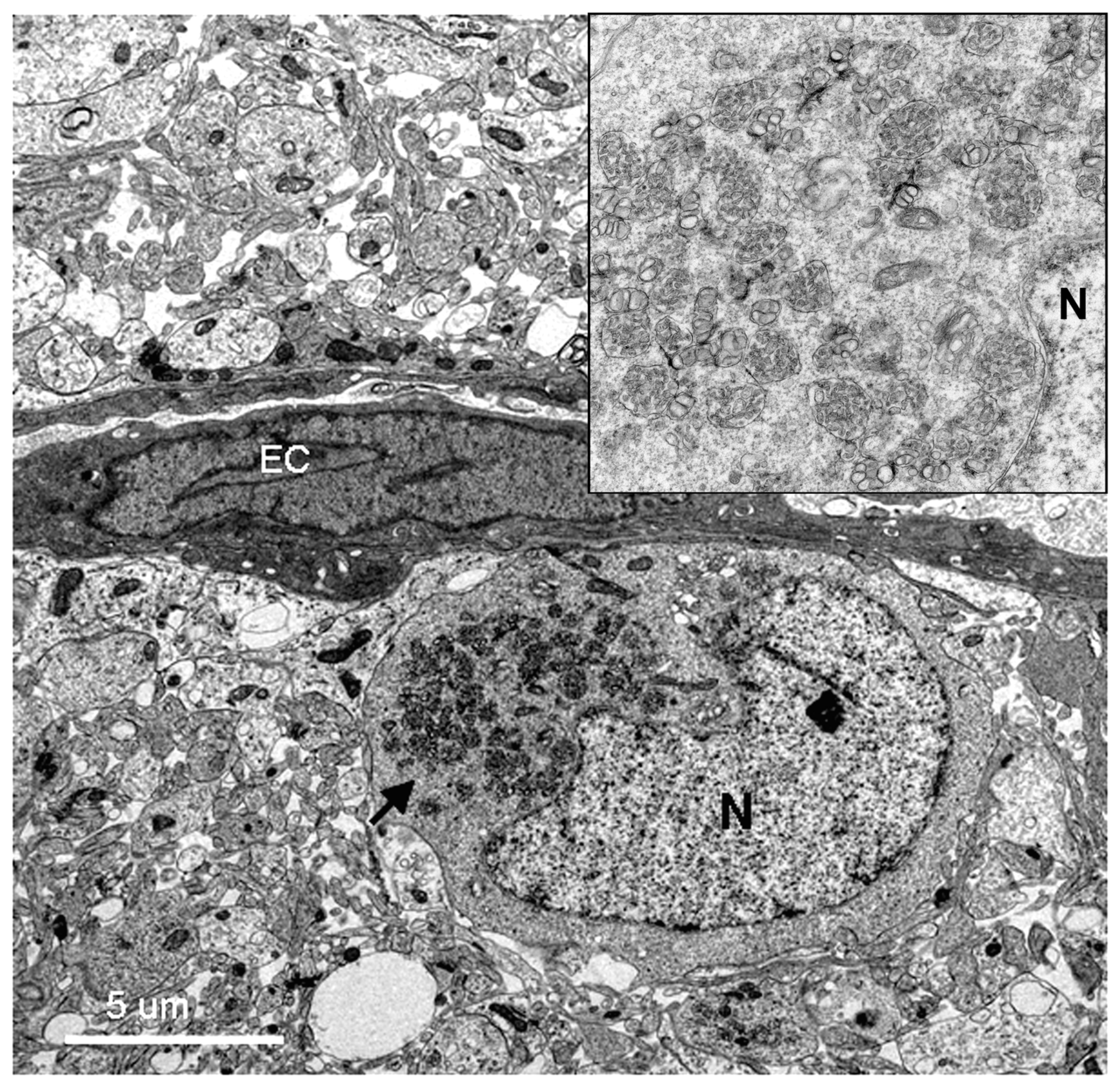
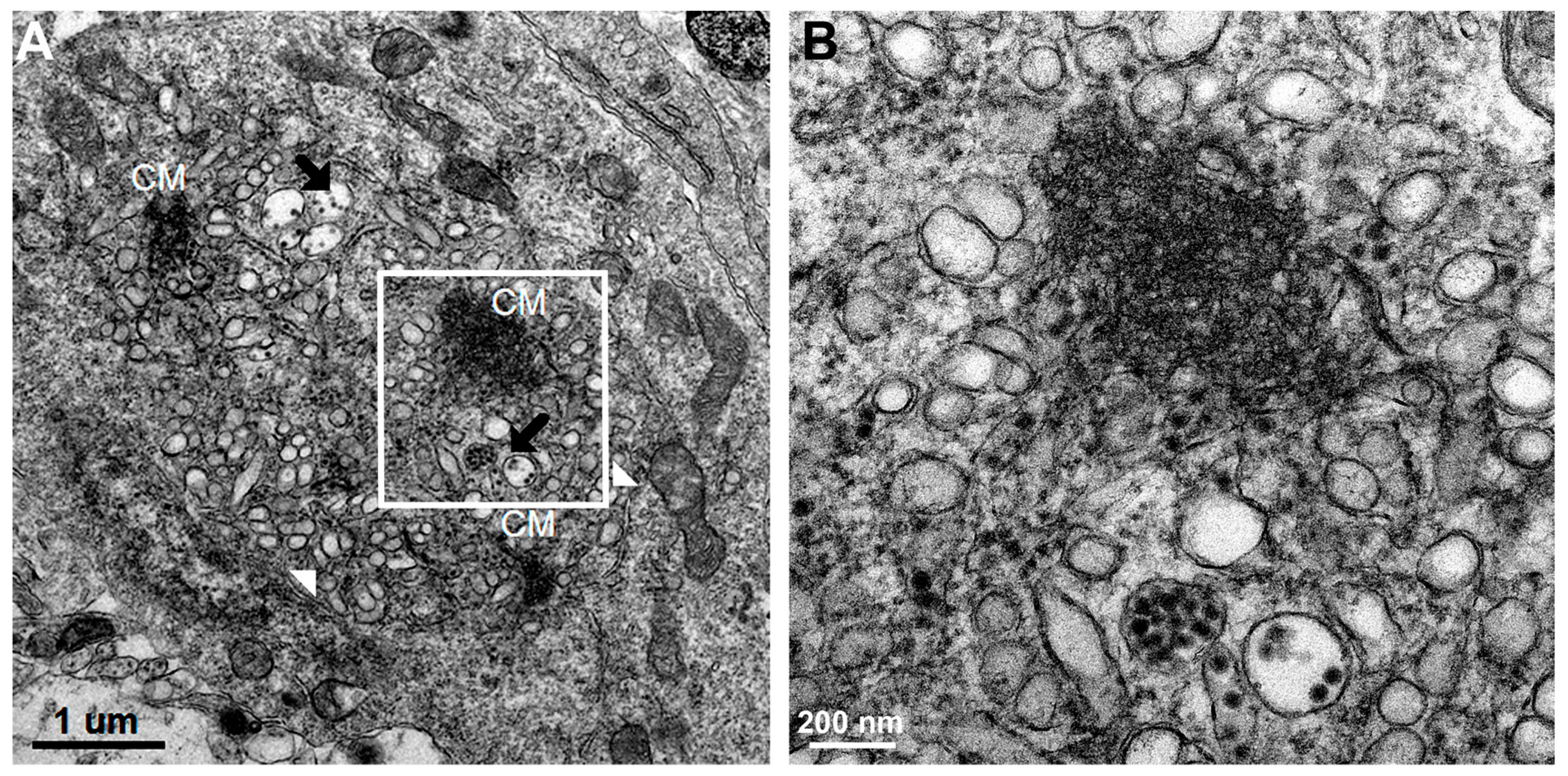
Using transmission electron microscopy, we confirmed all of the hallmarks of cytoplasmic membrane changes induced by DV infection of mouse neurons and microglial cells in the in vivo suckling mouse brain infection model. These features had recently been defined by electron tomography and modern cryo-preservative fixation applied to infections in mammalian and insect-cell cultures. The structures, when combined with associated functional studies as carried out by immuno-electron microscopic and molecular studies, gave a complete picture of the molecular mechanisms of how the replication and formation of new virions occurred (reviewed in [
3]). We also demonstrated some unique features that were found in the in vivo infections. First, there was a higher degree of membrane proliferative changes, with different shapes of intra-membranous vesicles ranging from rounded, oblong and tubular shapes, arranged in packets and distributed around the perinuclear area. Second, apart from newly formed virions with a size of 50 nm sizes and electron-dense nuclei, we also frequently found deformed and defective “virion-like vesicles” packed inside large vesicles. The latter features have not been previously described in in vitro infections. It was not known whether they were the result of excess but defective virus budding or if they were simply due to excessive induction of vesicles.
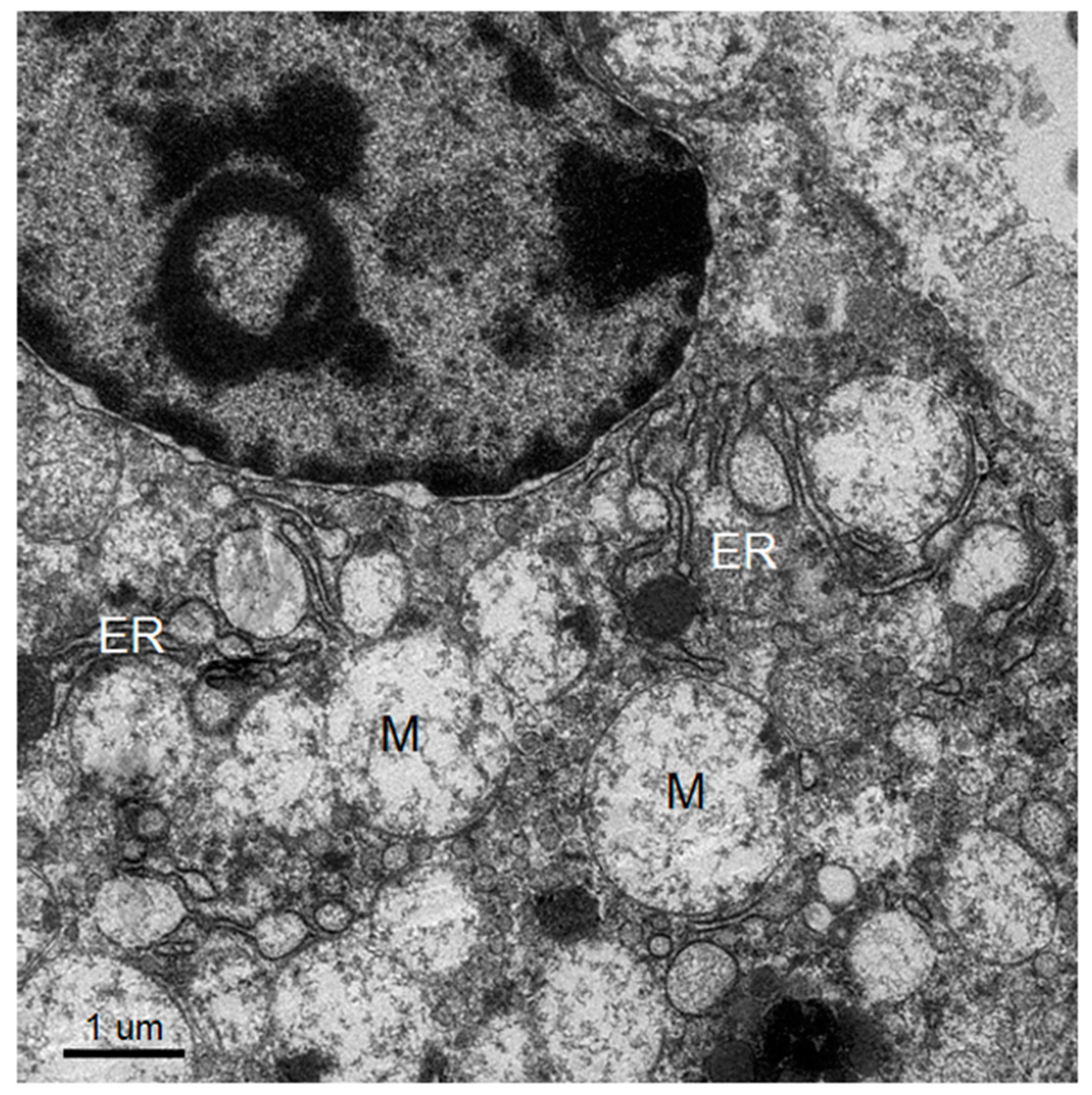
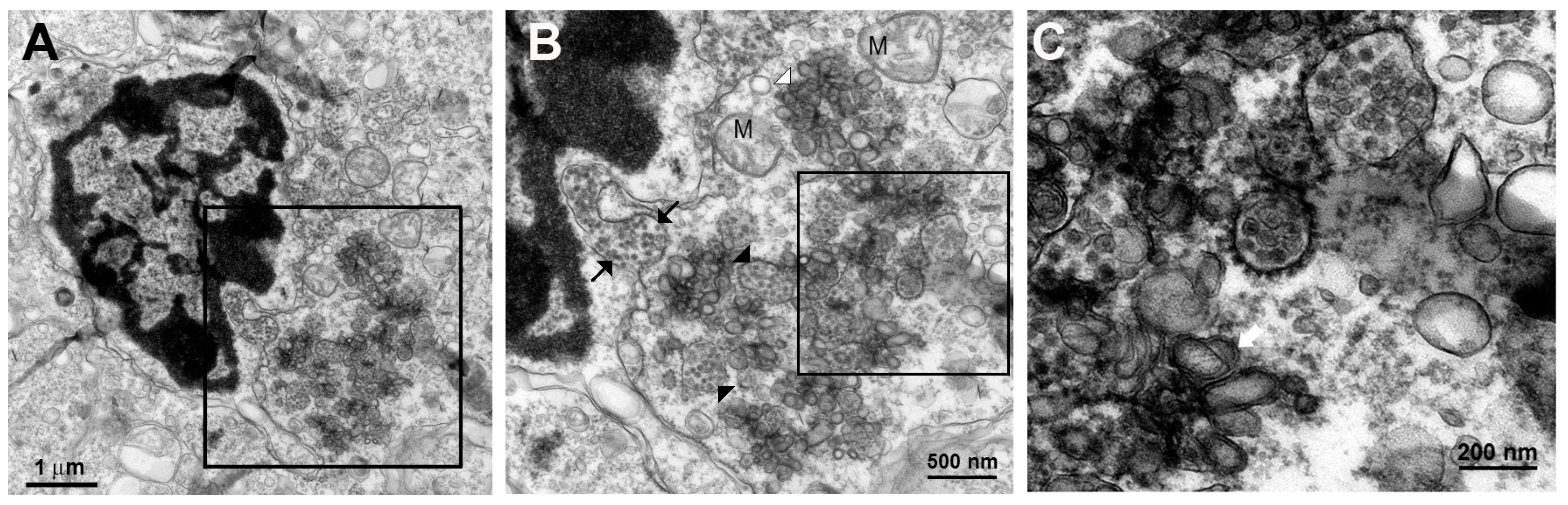
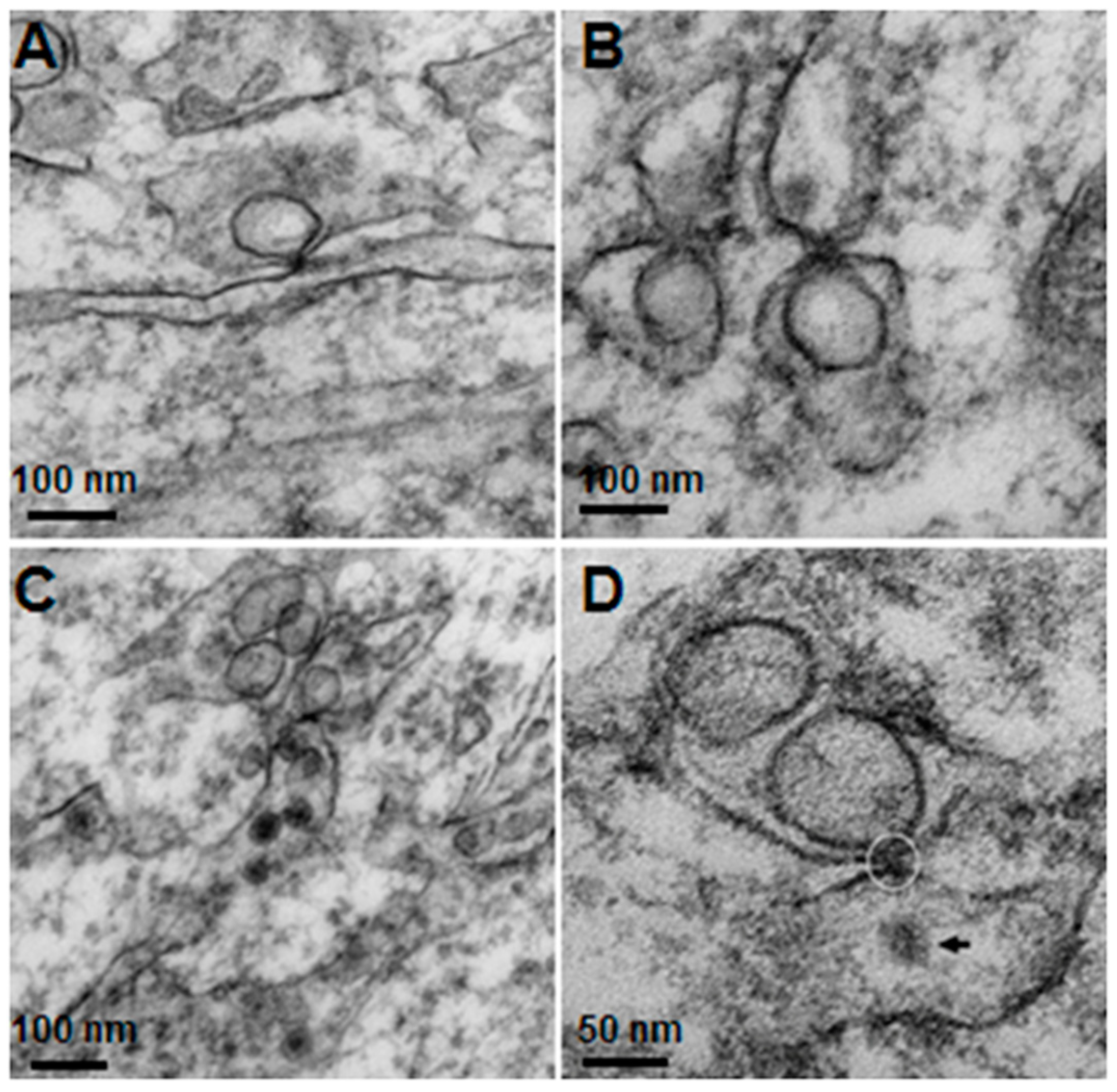
The knowledge of virus budding processes gained from the recent tomographic studies [
5] allowed us to search for similar structures and sites where the new virions are formed by budding in infected mouse brain. We could identify the particular areas where two opposing ER meet, forming an electron dense “spot”. On one side there is usually an intra-ER vesicle touching the spot of the opposing bare membrane of the neighboring ER (
Figure 5). According to the three-dimensional (3D) structure and tomographic studies [
5,
6], the site is the spot where the mouth of an invaginated vesicle containing the virus replicating complex meets the opposing ER membrane, delivering the positive RNA to the membrane and inducing the budding and formation of new virions in the ER cisternae. These spots were frequently found if carefully searched for newly formed virions that were usually found nearby.
Interestingly, despite the fact that there was extensive ER proliferation and Golgi structural changes facilitating viral replication, very few cell deaths and/or nuclear changes in neuronal and glial cells were observed at the time when the mice were completely paralyzed. Similar observations were made by Hase, et al. [
10] in the study of intracerebral infections of mouse brains by the closely related Japanese encephalitis virus (JEV). They commented that “the JEV does not appear to be strongly cytolytic, and it is conceivable that the cause of death for the mice is severe neuronal dysfunction rather than neuronal destruction”. This was in sharp contrast to our autopsy study in patients who died of severe dengue hemorrhagic fever. Severe liver cell damage was observed in the face of minimal replicative structural changes.
One obvious explanation was the long duration of virus infection in all patients, passing the active period of virus replication known to coincide with the early pyrexic phase of the disease. The ER structural changes might have therefore already been resolved. At the time of autopsy the patients had already been infected for a length of time, which included the symptomatic (4 to 11 days, see Materials and Methods) and the incubation periods. It has been documented that the incubation time of DHF varies widely from 4 to 10 days [
11,
12]; this means that the patients have been infected for a total length of 8 to 21 days. However, we were surprised by the rapidity of the disappearance and the paucity of DV-induced ER features, and the rare number of virions in the groups of patients who died early (only 4-5 days of illness,
Table 1). We would like to speculate that liver injury independent from virus infection (discussed below) might contribute to this acceleration. Without pathology information at the early phase of disease (which is not practical), it is difficult to gauge the contribution of liver to the total pool of virus in patients with different severities. The absence of ultrastructural evidence of virus replication in autopsy material has rendered the technology unsuitable for dengue virus diagnosis, unlike the immunohistochemistry and dengue RNA identification that are more sensitive and suitable.
Similar to this study, a key light microscopic feature of liver pathology reported in our former study, was the prominence of steatosis. Since steatosis has not been a feature shown in the in vitro DV infection of liver cell lines, this pointed to other processes. This is in accord with our previous report that there was no correlation between viral antigen staining (or the quantity of viral RNA found in tissues) and the degree of liver damage, suggesting that DV infection might not be the key contributor [
8]. Steatosis was the key pathologic feature in acute alcoholic and other non-alcoholic injuries such as tetracycline toxicity or in Reye’s syndrome [
13]. One of the hypotheses on the mechanism of liver damage was that the breakdown of the intestinal barrier allows bacterial pyrogens to reach the liver—a process also known as microbial translocation. Recent findings of raised blood levels of lipopolysaccharide in DHF patients, which correlated with disease severity, offered one possible factor that could injure the liver and suggested that the DV infection of the gut-associated-lymphoid tissues triggered microbial translocation injury to the liver [
14]. This speculation remains to be further investigated.
In conclusion, no virus-induced endoplasmic replicating structures have been identified in hepatocytes from DHF patients. Prominent findings were extensive cellular damage and steatosis. The latter process might accelerate the disappearance of the endoplasmic replicating structure occurred in early phase of the disease. It would be interesting to investigate in future study whether the structure could be identified in other organs found infected by the virus [
8]. In addition, this ultrastructural study of DV infection has shown for the first time that hallmark cytoplasmic features previously defined in in vitro DV-infected mammalian and insect cell models also occur in the in vivo DV-infected suckling mouse brain. This in vivo model can serve as an important tool for electron microscopists to explore human and experimental animal models to define organ tropism and the viral replication and/or pathological sites responsible for the pathogenesis of DHF and other closely related flaviviruses.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}