The Trypanosomal Transferrin Receptor of Trypanosoma Brucei—A Review


Abstract
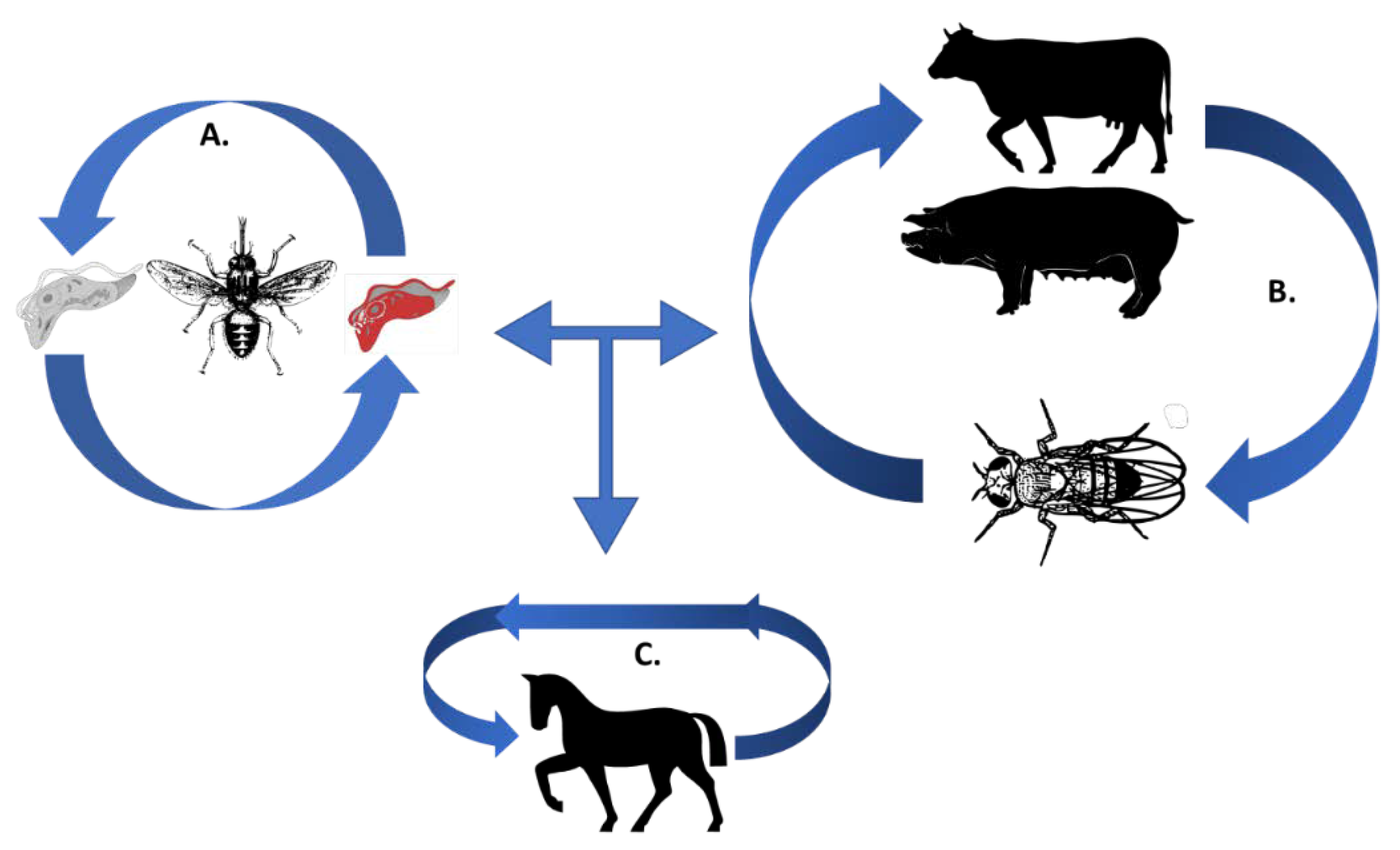
:1. Trypanosomes and Their Need for Iron during a Mammalian Infection
2. Expression Sites; the Trypanosomal Swiss Army Knife for Host Adaptation
3. The Trypanosomal Transferrin Receptor: A Structural Review
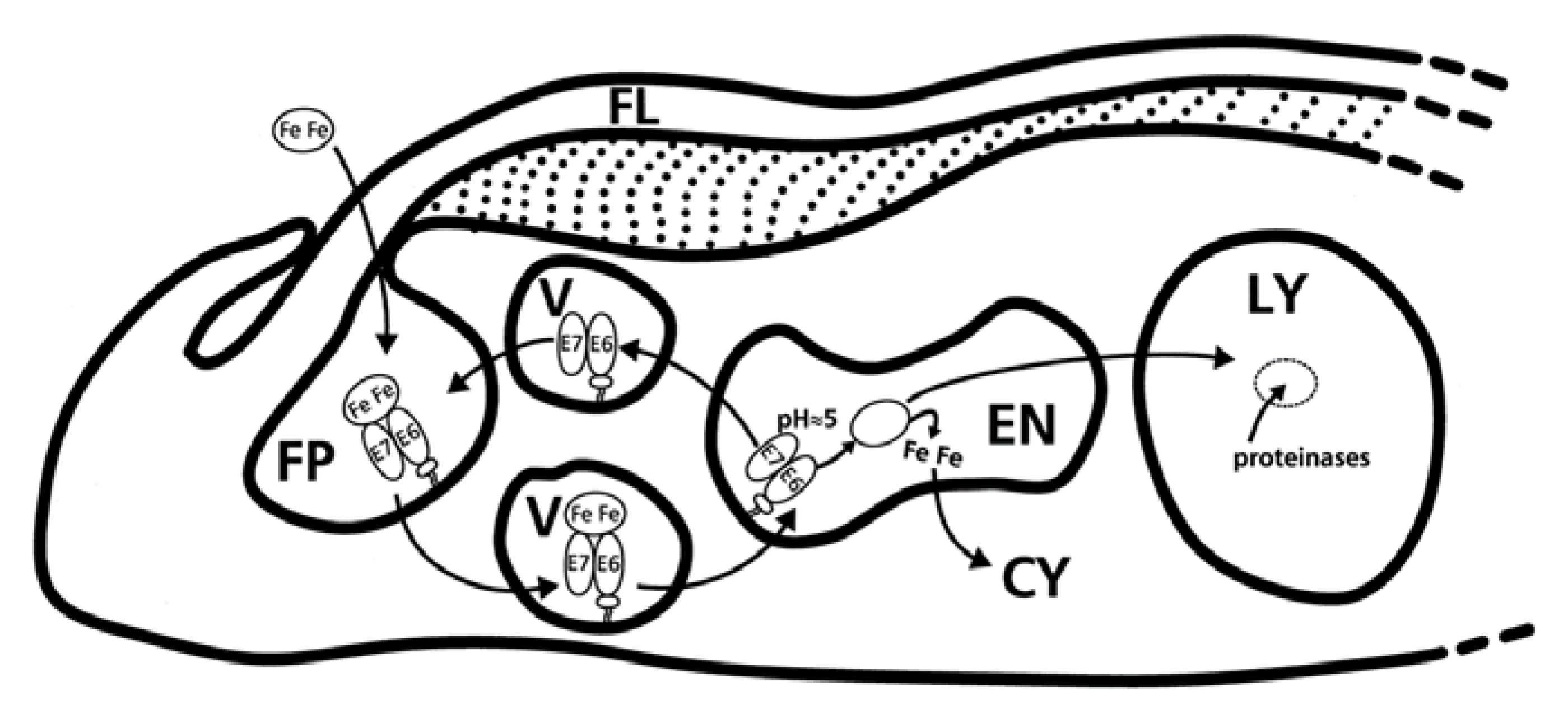
4. Fishing from a Hole; the Flagellar Pocket and the Quest for Iron
5. The Trypanosomal Transferrin Receptor as A Target for Chemotherapeutic Purposes
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aggett, P.J. Iron. In Present Knowledge in Nutrition; Erdman, J.W., Jr., Macdonald, I.A., Zeisel, S.H., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 506–520. ISBN 9780470959176. [Google Scholar]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aisen, P.; Listowsky, I. Iron Transport and Storage Proteins. Annu. Rev. Biochem. 1980, 49, 357–393. [Google Scholar] [CrossRef] [PubMed]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. Oxidation of tartartc acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Kosman, D.J. Iron metabolism in aerobes: Managing ferric iron hydrolysis and ferrous iron autoxidation. Coord. Chem. Rev. 2013, 100, 130–134. [Google Scholar] [CrossRef]
- Anderson, G.J.; Vulpe, C.D. Mammalian iron transport. Cell. Mol. Life Sci. 2009, 66, 3241–3261. [Google Scholar] [CrossRef]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 403–410. [Google Scholar] [CrossRef]
- Williams, J. The evolution of transferrin. Trends Biochem. Sci. 1982, 7, 394–397. [Google Scholar] [CrossRef]
- Morgan, E.H. Transferrin, biochemistry, physiology and clinical significance. Mol. Asp. Med. 1981, 4, 1–123. [Google Scholar] [CrossRef]
- Mizutani, K.; Toyoda, M.; Mikami, B. X-ray structures of transferrins and related proteins. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 203–211. [Google Scholar] [CrossRef]
- Reyes-López, M.; Piña-Vázquez, C.; Serrano-Luna, J. Transferrin: Endocytosis and Cell Signaling in Parasitic Protozoa. Biomed. Res. Int. 2015, 2015, 641392. [Google Scholar] [CrossRef] [PubMed]
- Macedo, M.F.; de Sousa, M. Transferrin and the transferrin receptor: Of magic bullets and other concerns. Inflamm. Allergy Drug Targets 2008, 7, 41–52. [Google Scholar] [CrossRef] [PubMed]
- OIE. OIE Animal Trypanosomoses. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals; OIE: Paris, France, 2018; Volume 91, pp. 399–404. ISBN 978-92-95108-18-9. [Google Scholar]
- Giordani, F.; Morrison, L.J.; Rowan, T.G.; De Koning, H.P.; Barrett, M.P. The animal trypanosomiases and their chemotherapy: A review. Parasitology 2016, 143, 1862–1889. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.J.; Vezza, L.; Rowan, T.; Hope, J.C. Animal African Trypanosomiasis: Time to Increase Focus on Clinically Relevant Parasite and Host Species. Trends Parasitol. 2016, 32, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vokaty, S.; Desquesnes, M. Proceedings of First Symposium on New World Trypanosomes; Vokaty, S., Desquesnes, M., Eds.; Biblioteca Orton IICA: Bridgetown, Barbados, 1999; ISBN 0253-4746. [Google Scholar]
- Achcar, F.; Kerkhoven, E.J.; Barrett, M.P. Trypanosoma brucei: Meet the system. Curr. Opin. Microbiol. 2014, 20, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Steverding, D. Low affinity of Trypanosoma brucei transferrin receptor to apotransferrin at pH 5 explains the fate of the ligand during endocytosis. FEBS Lett. 1996, 396, 87–89. [Google Scholar] [CrossRef]
- Thomson, R.; Genovese, G.; Canon, C.; Kovacsics, D.; Higgins, M.K.; Carrington, M.; Winkler, C.A.; Kopp, J.; Rotimi, C.; Adeyemo, A.; et al. Evolution of the primate trypanolytic factor APOL1. Proc. Natl. Acad. Sci. USA 2014, 111, E2130–E2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pays, E.; Vanhollebeke, B.; Vanhamme, L.; Paturiaux-Hanocq, F.; Nolan, D.P.; Pérez-Morga, D. The trypanolytic factor of human serum. Nat. Rev. Microbiol. 2006, 4, 477–486. [Google Scholar] [CrossRef]
- Currier, R.B.; Cooper, A.; Burrell-Saward, H.; MacLeod, A.; Alsford, S. Decoding the network of Trypanosoma brucei proteins that determines sensitivity to apolipoprotein-L1. PLoS Pathog. 2018, 14, e1006855. [Google Scholar] [CrossRef]
- Cooper, A.; Capewell, P.; Clucas, C.; Veitch, N.; Weir, W.; Thomson, R.; Raper, J.; MacLeod, A. A Primate APOL1 Variant That Kills Trypanosoma brucei gambiense. PLoS Negl. Trop. Dis. 2016, 10, e0004903. [Google Scholar] [CrossRef]
- Hajduk, S.L.; Moore, D.R.; Vasudevacharya, J.; Siqueira, H.; Torri, A.F.; Tytler, E.M.; Esko, J.D. Lysis of Trypanosoma brucei by a toxic subspecies of human high density lipoprotein. J. Biol. Chem. 1989, 264, 5210–5217. [Google Scholar] [PubMed]
- Radwanska, M.; Vereecke, N.; Deleeuw, V.; Pinto, J.; Magez, S. Salivarian Trypanosomosis: A Review of Parasites Involved, Their Global Distribution and Their Interaction with the Innate and Adaptive Mammalian Host Immune System. Front. Immunol. 2018, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Claes, F.; Büscher, P.; Touratier, L.; Goddeeris, B.M. Trypanosoma equiperdum: Master of disguise or historical mistake? Trends Parasitol. 2005, 21, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Touratier, L. Challenges of non-tsetse transmitted animal trypanosomoses (NTTAT). An outline and some perspectives. Ann. N. Y. Acad. Sci. 2000, 916, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Desquesnes, M.; Yangtara, S.; Kunphukhieo, P.; Jittapalapong, S.; Herder, S. Zoonotic trypanosomes in South East Asia: Attempts to control Trypanosoma lewisi using human and animal trypanocidal drugs. Infect. Genet. Evol. 2016, 44, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Pays, E.; Nolan, D.P. Expression and function of surface proteins in Trypanosoma brucei. Mol. Biochem. Parasitol. 1998, 91, 3–36. [Google Scholar] [CrossRef]
- David Barry, J.; McCulloch, R. Antigenic variation in trypanosomes: Enhanced phenotypic variation in a eukaryotic parasite. Adv. Parasitol. 2004, 49, 1–70. [Google Scholar] [CrossRef]
- Borst, P.; Frasch, A.C.C.; Bernards, A.; Hoeijmakers, J.H.; van der Ploeg, L.H.; Cross, G.A.M. The genes for variant antigens in trypanosomes. Am. J. Trop. Med. Hyg. 1980, 29, 1033–1036. [Google Scholar] [CrossRef]
- Pays, E.; Vanhamme, L.; Pérez-Morga, D. Antigenic variation in Trypanosoma brucei: Facts, challenges and mysteries. Curr. Opin. Microbiol. 2004, 7, 369–374. [Google Scholar] [CrossRef]
- Rudenko, G. African trypanosomes: The genome and adaptations for immune evasion. Essays Biochem. 2015, 51, 47–62. [Google Scholar] [CrossRef]
- Cross, G.A.M.; Kim, H.S.; Wickstead, B. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol. Biochem. Parasitol. 2014, 195, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V.; Barry, J.D. Expression site-associated genes transcribed independently of variant surface glycoprotein genes in Trypanosoma brucei. Mol. Biochem. Parasitol. 1991, 47, 31–41. [Google Scholar] [CrossRef]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The genome of the African trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Pays, E.; Vanhamme, L.; Pérez-Morga, D. Antigenic variation in Trypanosoma african: Memorandum. Bull. World Health Organ. 1978, 56, 389–401. [Google Scholar]
- Pays, E.; Vanhollebeke, B.; Uzureau, P.; Lecordier, L.; Pérez-Morga, D. The molecular arms race between African trypanosomes and humans. Nat. Rev. Microbiol. 2014, 12, 575–584. [Google Scholar] [CrossRef]
- Kassem, A.; Pays, E.; Vanhamme, L. Transcription is initiated on silent variant surface glycoprotein expression sites despite monoallelic expression in Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 2014, 111, 8943–8948. [Google Scholar] [CrossRef]
- Pays, E. The variant surface glycoprotein as a tool for adaptation in African trypanosomes. Microbes Infect. 2006, 8, 930–937. [Google Scholar] [CrossRef]
- Vanhamme, L.; Lecordier, L.; Pays, E. Control and function of the bloodstream variant surface glycoprotein expression sites in Trypanosoma brucei. Int. J. Parasitol. 2001, 31, 523–531. [Google Scholar] [CrossRef]
- Kolev, N.G.; Ramsdell, T.K.; Tschudi, C. Temperature shift activates bloodstream VSG expression site promoters in Trypanosoma brucei. Mol. Biochem. Parasitol. 2018, 226, 20–23. [Google Scholar] [CrossRef]
- Hertz-Fowler, C.; Figueiredo, L.M.; Quail, M.A.; Becker, M.; Jackson, A.; Bason, N.; Brooks, K.; Churcher, C.; Fahkro, S.; Goodhead, I.; et al. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Horn, D. Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 2014, 195, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pays, E.; Tebabi, P.; Pays, A.; Coquelet, H.; Revelard, P.; Salmon, D.; Steinert, M. The genes and transcripts of an antigen gene expression site from T. brucei. Cell 1989, 57, 835–845. [Google Scholar] [CrossRef]
- Vanhamme, L.; Postiaux, S.; Poelvoorde, P.; Pays, E. Differential regulation of ESAG transcripts in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 102, 35–42. [Google Scholar] [CrossRef]
- McCulloch, R.; Horn, D. What has DNA sequencing revealed about the VSG expression sites of african trypanosomes? Trends Parasitol. 2009, 25, 359–363. [Google Scholar] [CrossRef]
- Stockdale, C.; Swiderski, M.R.; Barry, J.D.; McCulloch, R. Antigenic variation in Trypanosoma brucei: Joining the DOTs. PLoS Biol. 2008, 6, 1386–1391. [Google Scholar] [CrossRef]
- Borst, P.; Bitter, W.; Blundell, P.A.; Chaves, I.; Cross, M.; Gerrits, H.; Van Leeuwen, F.; McCulloch, R.; Taylor, M.; Rudenko, G. Control of VSG gene expression sites in Trypanosoma brucei. Mol. Biochem. Parasitol. 1998, 91, 67–76. [Google Scholar] [CrossRef]
- Borst, P.; Fairlamb, A.H. Surface receptors and transporters of Trypanosoma brucei. Annu. Rev. Microbiol. 1998, 52, 745–778. [Google Scholar] [CrossRef]
- Pays, E.; Lips, S.; Nolan, D.; Vanhamme, L.; Pérez-Morga, D. The VSG expression sites of Trypanosoma brucei: Multipurpose tools for the adaptation of the parasite to mammalian hosts. Mol. Biochem. Parasitol. 2001, 114, 1–16. [Google Scholar] [CrossRef]
- Van Xong, H.; Vanhamme, L.; Chamekh, M.; Chimfwembe, C.E.; Van Den Abbeele, J.; Pays, A.; Van Melrvenne, N.; Hamers, R.; De Baetselier, P.; Pays, E. A VSG expression site-associated gene confers resistance to human serum in Trypanosoma rhodesiense. Cell 1998, 95, 839–846. [Google Scholar] [CrossRef]
- Lips, S.; Revelard, P.; Pays, E. Identification of a new expression site-associated gene in the complete 30.5 kb sequence from the AnTat 1.3A variant surface protein gene expression site of Trypanosoma brucei. Mol. Biochem. Parasitol. 1993, 62, 135–137. [Google Scholar] [CrossRef]
- Jackson, A.P.; Allison, H.C.; Barry, J.D.; Field, M.C.; Hertz-Fowler, C.; Berriman, M. A Cell-surface Phylome for African Trypanosomes. PLoS Negl. Trop. Dis. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The significance of transferrin receptor variation in Trypanosoma brucei. Trends Parasitol. 2003, 19, 125–127. [Google Scholar] [CrossRef]
- Nairz, M.; Dichtl, S.; Schroll, A.; Haschka, D.; Tymoszuk, P.; Theurl, I.; Weiss, G. Iron and innate antimicrobial immunity – Depriving the pathogen, defending the host. J. Trace Elem. Med. Biol. 2018, 48, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D.; Stierhof, Y.D.; Fuchs, H.; Tauber, R.; Overath, P. Transferrin-binding protein complex is the receptor for transferrin uptake in Trypanosoma brucei. J. Cell Biol. 1995, 131, 1173–1182. [Google Scholar] [CrossRef]
- Baral, T.N. Immunobiology of African trypanosomes: Need of alternative interventions. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Pays, E. Regulation of antigen gene expression in Trypanosoma brucei. Trends Parasitol. 2005, 21, 517–520. [Google Scholar] [CrossRef]
- Ansorge, I.; Steverding, D.; Melville, S.; Hartmann, C.; Clayton, C. Transcription of “inactive” expression sites in African trypanosomes leads to expression of multiple transferrin receptor RNAs in bloodstream forms. Mol. Biochem. Parasitol. 1999, 101, 81–94. [Google Scholar] [CrossRef]
- Mussmann, R.; Engstler, M.; Gerrits, H.; Kieft, R.; Toaldo, C.B.; Onderwater, J.; Koerten, H.; Van Luenen, H.G.A.M.; Borst, P. Factors affecting the level and localization of the transferrin receptor in Trypanosoma brucei. J. Biol. Chem. 2004, 279, 40690–40698. [Google Scholar] [CrossRef]
- Cully, D.F.; Ip, H.S.; Cross, G.A.M. Coordinate transcription of variant surface glycoprotein genes and an expression site associated gene family in Trypanosoma brucei. Cell 1985, 42, 173–182. [Google Scholar] [CrossRef]
- Wilson, M.E.; Britigan, B.E. Iron acquisition by parasitic protozoa. Parasitol. Today 1998, 14, 348–353. [Google Scholar] [CrossRef]
- Maier, A.; Steverding, D. Expression and purification of non-glycosylated Trypanosoma brucei transferrin receptor in insect cells. Exp. Parasitol. 2008, 120, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Salmon, D.; Geuskens, M.; Hanocq, F.; Hanocq-Quertier, J.; Nolan, D.; Ruben, L.; Pays, E. A novel heterodimeric transferrin receptor encoded by a pair of VSG expression site-associated genes in T. brucei. Cell 1994, 78, 75–86. [Google Scholar] [CrossRef]
- Salmon, D.; Hanocq-Quertier, J.; Paturiaux-Hanocq, F.; Pays, A.; Tebabi, P.; Nolan, D.P.; Michel, A.; Pays, E. Characterization of the ligand-binding site of the transferrin receptor in Trypanosoma brucei demonstrates a structural relationship with the N-terminal domain of the variant surface glycoprotein. EMBO J. 1997, 16, 7272–7278. [Google Scholar] [CrossRef] [PubMed]
- Gerrits, H.; Mußmann, R.; Bitter, W.; Kieft, R.; Borst, P. The physiological significance of transferrin receptor variations in Trypanosoma brucei. Mol. Biochem. Parasitol. 2002, 119, 237–247. [Google Scholar] [CrossRef]
- Salmon, D.; Paturiaux-Hanocq, F.; Poelvoorde, P.; Vanhamme, L.; Pays, E. Trypanosoma brucei: Growth differences in different mammalian sera are not due to the species-specificity of transferrin. Exp. Parasitol. 2005, 109, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Bitter, W.; Gerrits, H.; Kieft, R.; Borst, P. The role of transferrin-receptor variation in the host range of Trypanosoma brucei. Nature 1998, 391, 499–502. [Google Scholar] [CrossRef]
- Steverding, D. On the significance of host antibody response to the Trypanosoma brucei transferrin receptor during chronic infection. Microbes Infect. 2006, 8, 2777–2782. [Google Scholar] [CrossRef]
- Mehlert, A.; Bond, C.S.; Ferguson, M.A.J. The glycoforms of a Trypanosoma brucei variant surface glycoprotein and molecular modeling of a glycosylated surface coat. Glycobiology 2002, 12, 607–612. [Google Scholar] [CrossRef]
- Steverding, D.; Overath, P. Trypanosoma brucei with an active metacyclic variant surface gene expression site expresses a transferrin receptor derived from esag6 and esag7. Mol. Biochem. Parasitol. 1996, 78, 285–288. [Google Scholar] [CrossRef]
- Jensen, R.E.; Simpson, L.; Englund, P.T. What happens when Trypanosoma brucei leaves Africa. Trends Parasitol. 2008, 24, 425–428. [Google Scholar] [CrossRef]
- Mehlert, A.; Wormald, M.R.; Ferguson, M.A.J. Modeling of the N-glycosylated transferrin receptor suggests how transferrin binding can occur within the surface coat of trypanosoma brucei. PLoS Pathog. 2012, 8, e1002618. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef]
- Blum, M.L.; Down, J.A.; Gurnett, A.M.; Carrington, M.; Turner, M.J.; Wiley, D.C. A structural motif in the variant surface glycoproteins of Trypanosoma brucei. Nature 1993, 362, 603–609. [Google Scholar] [CrossRef]
- Bullen, J.J. The significance of iron in infection. Rev. Infect. Dis. 1981, 3, 1127–1138. [Google Scholar] [CrossRef]
- Bullen, J.J.; Rogers, H.J.; Griffiths, E. Role of Iron in Bacterial Infection. Curr. Top. Microbiol. Immunol. 2012, 80, 1–35. [Google Scholar] [CrossRef]
- Latunde-Dada, G.O. Iron metabolism: Microbes, mouse, and man. BioEssays 2009, 31, 1309–1317. [Google Scholar] [CrossRef]
- Stijlemans, B.; Beschin, A.; Magez, S.; Van Ginderachter, J.A.; De Baetselier, P. Iron Homeostasis and Trypanosoma brucei Associated Immunopathogenicity Development: A Battle/Quest for Iron. Biomed. Res. Int. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kořený, L.; Oborník, M.; Lukeš, J. Make It, Take It, or Leave It: Heme Metabolism of Parasites. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The transferrin receptor of Trypanosoma brucei. Parasitol. Int. 2000, 48, 191–198. [Google Scholar] [CrossRef]
- Steverding, D.; Sexton, D.W.; Chrysochoidi, N.; Cao, F. Trypanosoma brucei transferrin receptor can bind C-lobe and N-lobe fragments of transferrin. Mol. Biochem. Parasitol. 2012, 185, 99–105. [Google Scholar] [CrossRef]
- Coppens, I.; Opperdoes, F.R.; Courtoy, P.J.; Baudhuin, P. Receptor-Mediated Endocytosis in the Bloodstream Form of Trypanosoma brucei. J. Protozool. 1987, 34, 465–473. [Google Scholar] [CrossRef]
- Kabiri, M.; Steverding, D. Studies on the recycling of the transferrin receptor in Trypanosoma brucei using an inducible gene expression system. Eur. J. Biochem. 2000, 267, 3309–3314. [Google Scholar] [CrossRef]
- Higgins, M.K.; Lane-Serff, H.; MacGregor, P.; Carrington, A. Receptor’s Tale: An Eon in the Life of a Trypanosome Receptor. PLOS Pathog. 2017, 13, e1006055. [Google Scholar] [CrossRef]
- Gull, K. Host–parasite interactions and trypanosome morphogenesis: A flagellar pocketful of goodies. Curr. Opin. Microbiol. 2003, 6, 365–370. [Google Scholar] [CrossRef]
- Landfear, S.M.; Ignatushchenko, M. The flagellum and flagellar pocket of trypanosomatids. Mol. Biochem. Parasitol. 2001, 115, 1–17. [Google Scholar] [CrossRef]
- Perdomo, D.; Bonhivers, M.; Robinson, D.R. The Trypanosome Flagellar Pocket Collar and Its Ring Forming Protein-TbBILBO1. Cells 2016, 5, 9. [Google Scholar] [CrossRef]
- Ligtenberg, M.J.; Bitter, W.; Kieft, R.; Steverding, D.; Janssen, H.; Calafat, J.; Borst, P. Reconstitution of a surface transferrin binding complex in insect form Trypanosoma brucei. EMBO J. 1994, 13, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Carrington, M. The trypanosome flagellar pocket. Nat. Rev. Microbiol. 2009, 7, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Goulding, D.; Field, M.C. Clathrin-mediated endocytosis is essential in Trypanosoma brucei. EMBO J. 2003, 22, 4991–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanya, S.; Mensa-Wilmot, K. Diacylglycerol-stimulated endocytosis of transferrin in trypanosomatids is dependent on tyrosine kinase activity. PLoS ONE 2010, 5, e8538. [Google Scholar] [CrossRef]
- Subramanya, S.; Hardin, F.C.; Steverding, D.; Mensa-Wilmot, K. Glycosylphosphatidylinositol-specific phospholipase C regulates transferrin endocytosis in the African trypanosome. Biochem. J. 2009, 417, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Subramanya, S.; Mensa-Wilmot, K. Regulated cleavage of intracellular glycosylphosphatidylinositol in a trypanosome: Peroxisome-to-endoplasmic reticulum translocation of a phospholipase C. FEBS J. 2006, 273, 2110–2126. [Google Scholar] [CrossRef]
- Mackey, Z.B.; O’Brien, T.C.; Greenbaum, D.C.; Blank, R.B.; McKerrow, J.H. A cathepsin B-like protease is required for host protein degradation in Trypanosoma brucei. J. Biol. Chem. 2004, 279, 48426–48433. [Google Scholar] [CrossRef]
- O’Brien, T.C.; Mackey, Z.B.; Fetter, R.D.; Choe, Y.; O’Donoghue, A.J.; Zhou, M.; Craik, C.S.; Caffrey, C.R.; McKerrow, J.H. A parasite cysteine protease is key to host protein degradation and iron acquisition. J. Biol. Chem. 2008, 283, 28934–28943. [Google Scholar] [CrossRef]
- Hall, B.S.; Smith, E.; Langer, W.; Jacobs, L.A.; Goulding, D.; Field, M.C. Developmental variation in Rab11-dependent trafficking in Trypanosoma brucei. Eukaryot. Cell 2005, 4, 971–980. [Google Scholar] [CrossRef]
- Steverding, D. Bloodstream forms of Trypanosoma brucei require only small amounts of iron for growth. Parasitol. Res. 1998, 84, 59–62. [Google Scholar] [CrossRef]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.C.; Mclatchie, A.P.; Kelly, J.M. Evidence that transport of iron from the lysosome to the cytosol in African trypanosomes is mediated by a mucolipin orthologue. Mol. Microbiol. 2013, 89, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Horáková, E.; Lukeš, J. Iron-associated biology of Trypanosoma brucei. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Mußmann, R.; Janssen, H.; Calafat, J.; Engstler, M.; Ansorge, I.; Clayton, C.; Borst, P. The expression level determines the surface distribution of the transferrin receptor in Trypanosoma brucei. Mol. Microbiol. 2003, 47, 23–35. [Google Scholar] [CrossRef]
- Van Luenen, H.G.A.M.; Kieft, R.; Mußmann, R.; Engstler, M.; Ter Riet, B.; Borst, P. Trypanosomes change their transferrin receptor expression to allow effective uptake of host transferrin. Mol. Microbiol. 2005, 58, 151–165. [Google Scholar] [CrossRef]
- Benz, C.; Lo, W.; Fathallah, N.; Connor-Guscott, A.; Benns, H.J.; Urbaniak, M.D. Dynamic regulation of the Trypanosoma brucei transferrin receptor in response to iron starvation is mediated via the 3’UTR. PLoS ONE 2018, 441931. [Google Scholar] [CrossRef]
- Tiengwe, C.; Bush, P.J.; Bangs, J.D. Controlling transferrin receptor trafficking with GPI-valence in bloodstream stage African trypanosomes. PLoS Pathog. 2017, 13, e1006366. [Google Scholar] [CrossRef]
- Stijlemans, B.; Brys, L.; Korf, H.; Bieniasz-Krzywiec, P.; Sparkes, A.; Vansintjan, L.; Leng, L.; Vanbekbergen, N.; Mazzone, M.; Caljon, G.; et al. MIF-Mediated Hemodilution Promotes Pathogenic Anemia in Experimental African Trypanosomosis. PLOS Pathog. 2016, 12, e1005862. [Google Scholar] [CrossRef]
- Naessens, J. Bovine trypanotolerance: A natural ability to prevent severe anaemia and haemophagocytic syndrome? Int. J. Parasitol. 2006, 36, 521–528. [Google Scholar] [CrossRef]
- Coffey, R.; Ganz, T. Iron homeostasis: An anthropocentric perspective. J. Biol. Chem. 2017, 292, 12727–12734. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.C.; Kelly, J.M. Iron metabolism in trypanosomatids, and its crucial role in infection. Parasitology 2010, 137, 899–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, P.; Tripathi, A.K.; Clark, M.A.; Hand, C.C.; Rienhoff, H.Y.; Sullivan, D.J. Antimalarial iron chelator, FBS0701, shows asexual and gametocyte Plasmodium falciparum activity and single oral dose cure in a murine malaria model. PLoS ONE 2012, 7, e37171. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Siddiqui, A.A.; Saha, S.J.; De, R.; Mazumder, S.; Banerjee, C.; Iqbal, M.S.; Nag, S.; Adhikari, S.; Bandyopadhyay, U. Antimalarial activity of small-molecule benzothiazole hydrazones. Antimicrob. Agents Chemother. 2016, 60, 4217–4228. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.S.; Pinto, E.G.; Steverding, D.; Pleban, K.; Tempone, A.G.; Hider, R.C.; Wagner, G.K. Conjugation to 4-aminoquinoline improves the anti-trypanosomal activity of Deferiprone-type iron chelators. Bioorganic Med. Chem. 2013, 21, 805–813. [Google Scholar] [CrossRef]
- Bergeron, R.J.; Wiegand, J.; Bharti, N.; McManis, J.S.; Singh, S. Desferrithiocin analogue iron chelators: Iron clearing efficiency, tissue distribution, and renal toxicity. BioMetals 2011, 24, 239–258. [Google Scholar] [CrossRef]
- Rienhoff, H.Y.; Viprakasit, V.; Tay, L.; Harmatz, P.; Vichinsky, E.; Chirnomas, D.; Kwiatkowski, J.L.; Tapper, A.; Kramer, W.; Porter, J.B.; et al. A phase 1 dose-escalation study: Safety, tolerability, and pharmacokinetics of FBS0701, a novel oral iron chelator for the treatment of transfusional iron overload. Haematologica 2011, 96, 521–525. [Google Scholar] [CrossRef]
- Merschjohann, K.; Steverding, D. In vitro growth inhibition of bloodstream forms of Trypanosoma brucei and Trypanosoma congolense by iron chelators. Kinetoplastid Biol. Dis. 2006, 5, 3. [Google Scholar] [CrossRef]
- Tyler, K.M.; Higgs, P.G.; Matthews, K.R.; Gull, K. Limitation of Trypanosoma brucei parasitaemia results from density-dependent parasite differentiation and parasite killing by the host immune response. Proc. R. Soc. B Biol. Sci. 2001, 268, 2235–2243. [Google Scholar] [CrossRef]
- Muyldermans, S.; Vincke, C. Structure and Function of Camelid VHH. Encycl. Immunobiol. 2016, 2, 153–159. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient Cancer Therapy with a Nanobody-Based Conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar] [CrossRef] [Green Version]
- Conrath, K.E.; Lauwereys, M.; Galleni, M.; Matagne, A.; Frere, J.M.; Kinne, J.; Wyns, L.; Muyldermans, S. Beta-Lactamase Inhibitors Derived from Single-Domain Antibody Fragments Elicited in the Camelidae. Antimicrob. Agents Chemother. 2001, 45, 2807–2812. [Google Scholar] [CrossRef] [PubMed]
- Baral, T.N.; Magez, S.; Stijlemans, B.; Conrath, K.; Vanhollebeke, B.; Pays, E.; Muyldermans, S.; De Baetselier, P. Experimental therapy of African trypanosomiasis with a nanobody-conjugated human trypanolytic factor. Nat. Med. 2006, 12, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Stijlemans, B.; Conrath, K.; Cortez-Retamozo, V.; Van Xong, H.; Wyns, L.; Senter, P.; Revets, H.; De Baetselier, P.; Muyldermans, S.; Magez, S. Efficient Targeting of Conserved Cryptic Epitopes of Infectious Agents by Single Domain Antibodies: AFRICAN TRYPANOSOMES AS PARADIGM. J. Biol. Chem. 2004, 279, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Stijlemans, B.; Caljon, G.; Natesan, S.K.; Saerens, D.; Conrath, K.; Pérez-Morga, D.; Skepper, J.N.; Nikolaou, A.; Brys, L.; Pays, E.; et al. High affinity nanobodies against the Trypanosome brucei VSG are potent trypanolytic agents that block endocytosis. PLoS Pathog. 2011, 7, e1002072. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel Single-domain Antibodies as Modular Building Units in Bispecific and Bivalent Antibody Constructs. J. Biol. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, Á.M.; Sainz-Pastor, N.; Bonet, J.; Oliva, B.; Álvarez-Vallina, L. Multivalent antibodies: When design surpasses evolution. Trends Biotechnol. 2010, 28, 355–362. [Google Scholar] [CrossRef]
- Hmila, I.; Abdallah R, B.A.B.; Saerens, D.; Benlasfar, Z.; Conrath, K.; El Ayeb, M.; Muyldermans, S.; Bouhaouala-Zahar, B. VHH, bivalent domains and chimeric Heavy chain-only antibodies with high neutralizing efficacy for scorpion toxin AahI’. Mol. Immunol. 2008, 45, 3847–3856. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Trypanosome TfR | Human TfR |
|---|---|---|
| Subunit organization | Heterodimer of ESAG6 (50–60 kDa) and ESAG7 (40–42 kDa) | Homodimer of 90-kDa subunits |
| Post-translational modifications | ESAG6: 2–5 N-linked glycans ESAG7: 2–3 N-linked glycans | Per subunit: 3 N-linked glycan 1 Phosphorylation (Ser 24) 1 Acylation (Cys 62) |
| Membrane anchorage | GPI-anchor at the C-terminus of ESAG6 | 1 Transmembrane domain per subunit |
| Copy number per cell | 3000 | 20000–700000 |
| Ligand/receptor stoichiometry | 1 Transferrin molecule per heterodimer | 1 Transferrin molecule per monomer |
| Transferrin Receptor (nM) | Transferrin | pH | Kd-Value | Reference |
|---|---|---|---|---|
| T. brucei strain 427 | Holo bovine | 7 | 2.6–3.6 | [57,69] |
| 5 | 12 | [19] | ||
| Apo bovine | 7 | 20 | [103] | |
| 5 | 1100 | [19] | ||
| Human cells | Holo human | 7 | 1.9–7.7 | [104] |
| 5 | 13 | [104] | ||
| Apo human | 7 | >700 | [104] | |
| 5 | 13–21 | [104] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kariuki, C.K.; Stijlemans, B.; Magez, S. The Trypanosomal Transferrin Receptor of Trypanosoma Brucei—A Review. Trop. Med. Infect. Dis. 2019, 4, 126. https://doi.org/10.3390/tropicalmed4040126
Kariuki CK, Stijlemans B, Magez S. The Trypanosomal Transferrin Receptor of Trypanosoma Brucei—A Review. Tropical Medicine and Infectious Disease. 2019; 4(4):126. https://doi.org/10.3390/tropicalmed4040126
Chicago/Turabian StyleKariuki, Christopher K., Benoit Stijlemans, and Stefan Magez. 2019. "The Trypanosomal Transferrin Receptor of Trypanosoma Brucei—A Review" Tropical Medicine and Infectious Disease 4, no. 4: 126. https://doi.org/10.3390/tropicalmed4040126
APA StyleKariuki, C. K., Stijlemans, B., & Magez, S. (2019). The Trypanosomal Transferrin Receptor of Trypanosoma Brucei—A Review. Tropical Medicine and Infectious Disease, 4(4), 126. https://doi.org/10.3390/tropicalmed4040126