The Drugs of Sleeping Sickness: Their Mechanisms of Action and Resistance, and a Brief History


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
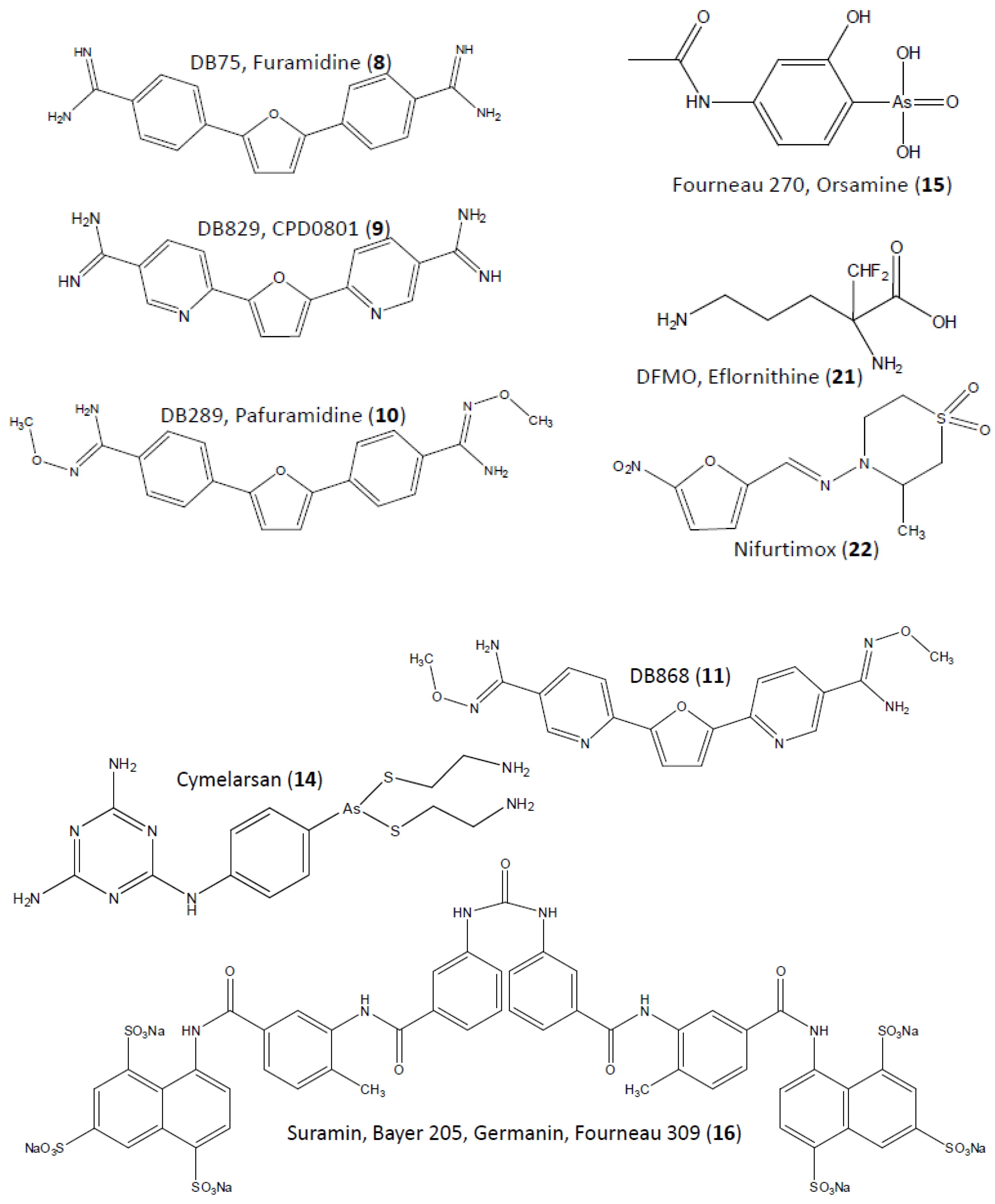
2. Diamidines
3. Melaminophenyl Arsenicals
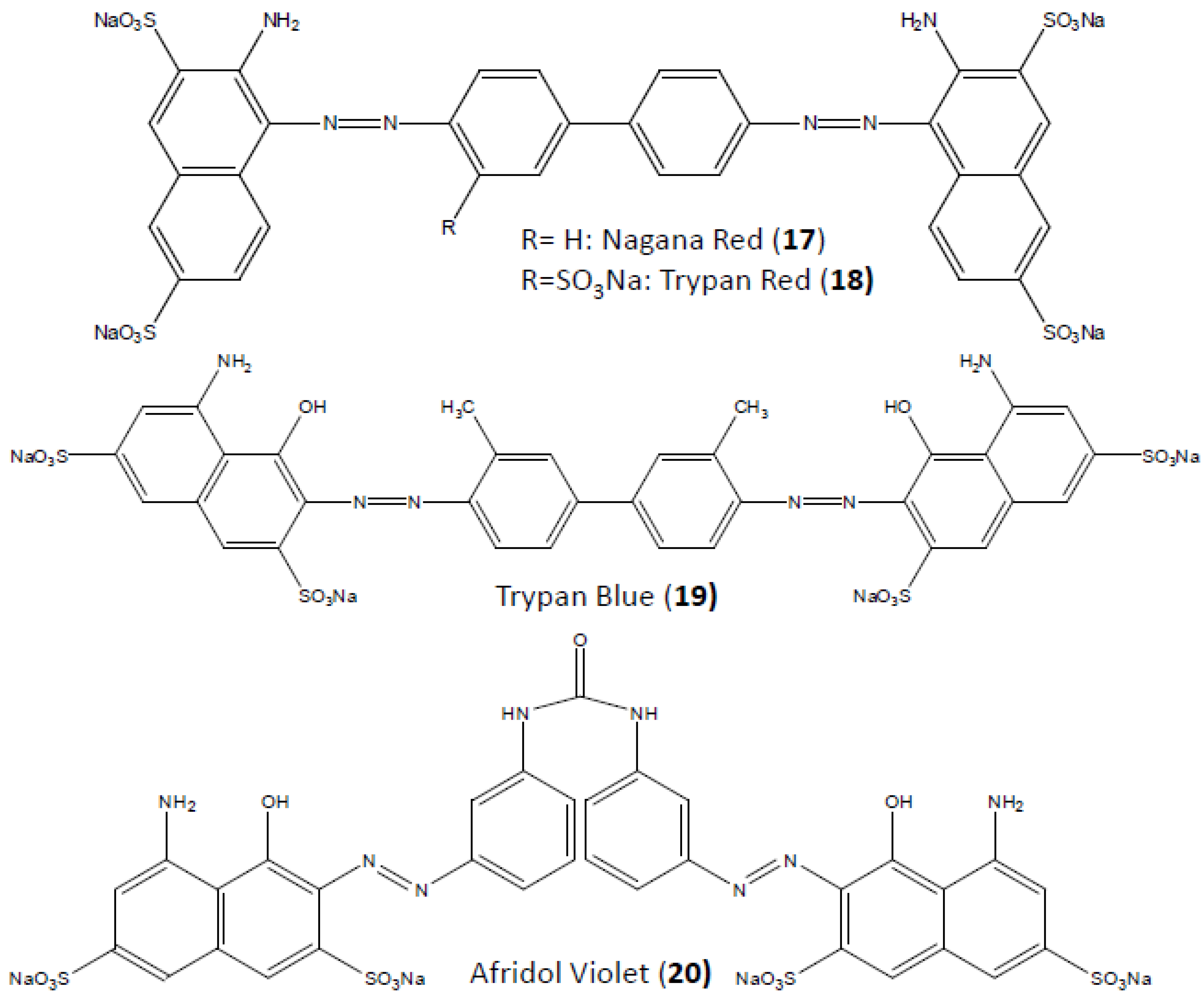
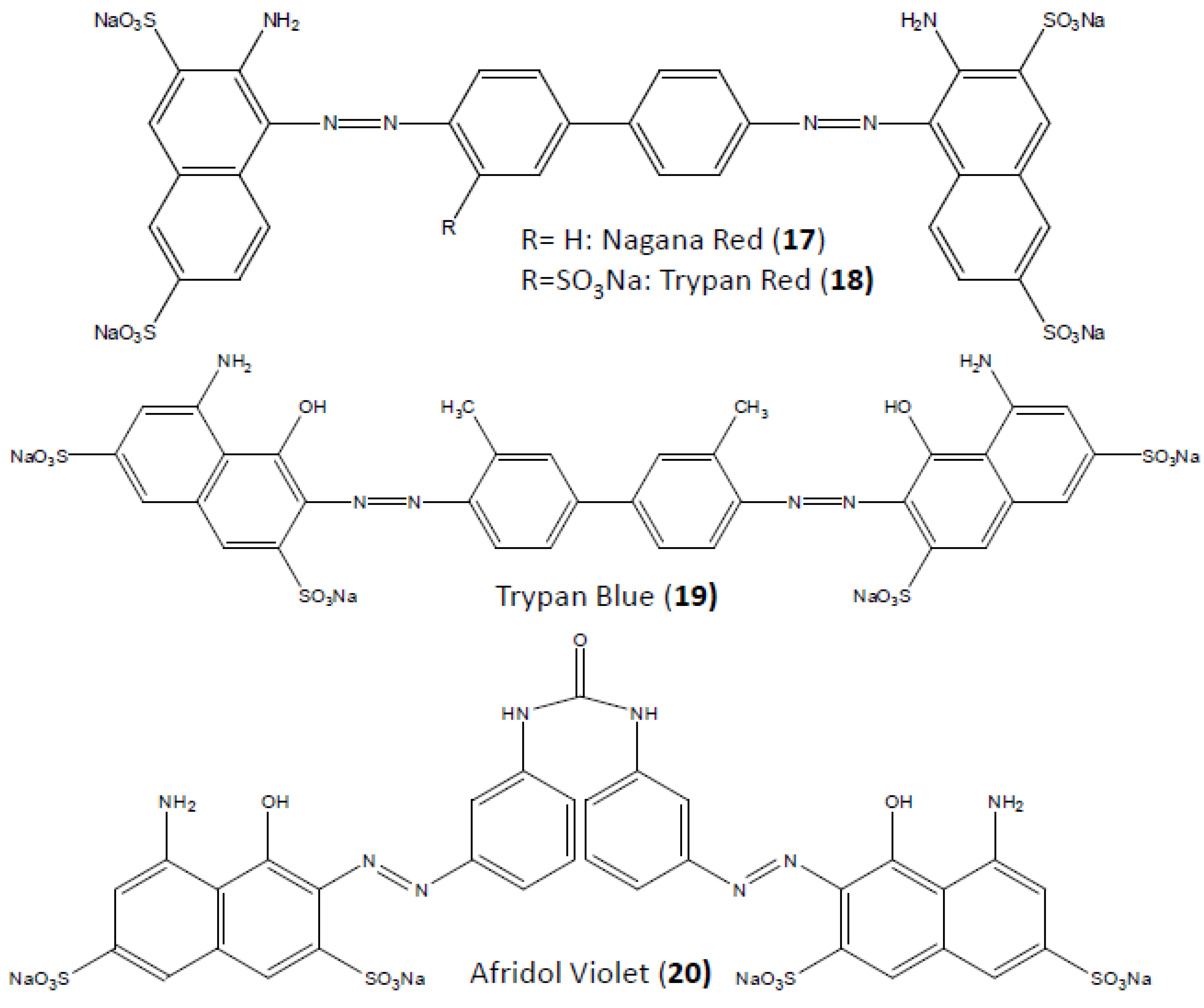
4. Suramin
5. Eflornithine
6. Nifurtimox
7. A Perspective on Drug Resistance in African Trypanosomiasis
Conflicts of Interest
References
- Fairlamb, A.H. Fexinidazole for the treatment of human African trypanosomiasis. Drugs Today 2019, 55, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Eperon, G.; Balasegaram, M.; Potet, J.; Mowbray, C.; Valverde, O.; Chappuis, F. Treatment options for second-stage gambiense human African trypanosomiasis. Expert Rev. Anti-Infect. Ther. 2014, 12, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- R&D Portfolio Update February 2019: DNDi Sleeping Sickness Programme. Available online: www.dndi.org/2019/media-centre/news-views-stories/news/sleepingsickness_rnd_status_2019/ (accessed on 15 December 2019).
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, A. Mort par hypoglycémie dans les trypanosomiases aiguës. CR Soc. Biol. 1928, 99, 656–657. [Google Scholar]
- Nieman, R.E.; Kelly, J.J.; Waskin, H.A. Severe African trypanosomiasis with spurious hypoglycemia. J. Infect. Dis. 1989, 159, 360–362. [Google Scholar] [CrossRef]
- Williamson, J. Review of chemotherapeutic and chemoprophylactic drugs. In The African Trypanosomiases; Mulligan, H.W., Ed.; George Allen/Unwin Ltd.: London, UK, 1970; pp. 125–221. [Google Scholar]
- Schern, K.; Artagaveytia-Allende, R. Zur glykopriven Therapie und Prophylaxe mit sowohl toxische als auch atoxische wirkende Substanzen bei der experimentellen Trypanosomen-und Treponemen-Infektion. Z Immun. Exp. Ther. 1936, 89, 21–64. [Google Scholar]
- Von Janscó, N.; Von Janscó, H. Chemotherapeutische Wirkung und Kohlehydratstoffwechsel: Die Heilwirkung von Guanidinderivaten auf die Trypanosomeninfektion. Z Immun. Exp. Ther. 1935, 86, 1–30. [Google Scholar]
- King, H.; Lourie, E.M.; Yorke, W. New trypanocidal substances. Lancet 1937, 223, 1360–1363. [Google Scholar] [CrossRef]
- Lourie, E.M.; Yorke, W. Studies in Chemotherapy XVI: The trypanocidal action of synthalin. Ann. Trop. Med. Parasitol. 1937, 31, 435–445. [Google Scholar] [CrossRef]
- Lourie, E.M.; Yorke, W. Studies in Chemotherapy XVI: The trypanocidal action of certain aromatic diamidines. Ann. Trop. Med. Parasitol. 1939, 33, 289–304. [Google Scholar] [CrossRef]
- Napier, L.E.; Sen Gupta, P.C. A peculiar neurological sequel to administration of 4:400-diamidino-diphenyl-ethylene (M&B 744). Indian Med. Gaz. 1942, 77, 71–74. [Google Scholar]
- Friedheim, E.A.H. MelB in the treatment of human trypanosomiasis. Am. J. Trop. Med. 1949, 29, 173–180. [Google Scholar] [CrossRef]
- Temu, S. Summary of cases of human early trypanosomiasis treated with Berenil at EATRO. Trans. R. Soc. Trop. Med. Hyg. 1975, 69, 277. [Google Scholar]
- Lansiaux, A.; Tanious, F.; Mishal, Z.; Dassonneville, L.; Kumar, A.; Stephens, C.E.; Hu, Q.; Wilson, W.D.; Boykin, D.W.; Bailly, C. Distribution of furamidine analogues in tumor cells: Targeting of the nucleus or mitochondria depending on the amidine substitution. Cancer Res. 2002, 62, 7219–7229. [Google Scholar] [PubMed]
- Mathis, A.M.; Bridges, A.S.; Ismail, M.A.; Kumar, A.; Francesconi, I.; Anbazhagan, M.; Hu, Q.; Tanious, F.A.; Wenzler, T.; Saulter, J.; et al. Diphenyl furans and aza analogs: Effects of structural modification on in vitro activity, DNA binding, and accumulation and distribution in trypanosomes. Antimicrob. Agents Chemother. 2007, 51, 2801–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, B.; Lee, M.P.H.; Hamelberg, D.; Joubert, A.; Bailly, C.; Brun, R.; Neidle, S.; Wilson, W.D. Strong binding in the DNA minor groove by an aromatic diamidine with a shape that does not match the curvature of the groove. J. Am. Chem. Soc. 2002, 124, 13680–13681. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.D.; Tanious, F.A.; Mathis, A.; Tevis, D.; Hall, J.E.; Boykin, D.W. Antiparasitic compounds that target DNA. Biochimie 2008, 90, 999–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wang, F.; Tong, Y.; Li, L.-F.; Liu, Y.; Gao, W.-Q. Pentamidine inhibits prostate cancer progression via selectively inducing mitochondrial DNA depletion and dysfunction. Cell Prolif. 2019, e12718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, M.L.; Krishna, S.; Burchmore, R.J.S.; Brun, R.; De Koning, H.P.; Boykin, D.W.; Tidwell, R.R.; Hall, J.E.; Barrett, M.P. Detection of arsenical drug resistance in Trypanosoma brucei using a simple fluorescence test. Lancet 2005, 366, 486–487. [Google Scholar] [CrossRef]
- Damper, D.; Patton, C.L. Pentamidine transport and sensitivity in brucei-group trypanosomes. J Protozool 1976, 23, 39–356. [Google Scholar] [CrossRef]
- Damper, D.; Patton, C.L. Pentamidine transport in Trypanosoma brucei—Kinetics and specificity. Biochem. Pharmacol. 1976, 25, 271–276. [Google Scholar] [CrossRef]
- Mathis, A.M.; Holman, J.L.; Sturk, L.M.; Ismail, M.A.; Boykin, D.W.; Tidwell, R.R.; Hall, J.E. Accumulation and intracellular distribution of antitrypanosomal diamidine compounds DB75 and DB820 in African trypanosomes. Antimicrob. Agents Chemother. 2006, 50, 2185–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eze, A.A.; Gould, M.K.; Munday, J.C.; Tagoe, D.N.A.; Stelmanis, V.; Schnaufer, A.; De Koning, H.P. Loss of mitochondrial membrane potential is a late adaptation of Trypanosoma brucei brucei to isometamidium preceded by mutations in the γ subunit of the F1F0-ATPase. PLoS Negl. Trop. Dis. 2016, 10, e0004791. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.M.; Al-Salabi, M.I.; El Sabbagh, N.; Quashie, N.B.; Alkhaldi, A.A.; Escale, R.; Smith, T.K.; Vial, H.J.; De Koning, H.P. Symmetrical choline-derived dications display strong anti-kinetoplastid activity. J. Antimicrob. Chemother. 2011, 66, 111–125. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.P.; Wong, P.E.; Burchmore, R.J.; De Koning, H.P.; Barrett, M.P. Trypanocidal furamidine analogues: Influence of pyridine nitrogens on trypanocidal activity, transport kinetics and resistance patterns. Antimicrob. Agents Chemother. 2011, 55, 2352–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanteri, C.A.; Tidwell, R.R.; Meshnick, S.R. The mitochondrion is a site of trypanocidal action of the aromatic diamidine DB75 in bloodstream forms of Trypanosoma brucei. Antimicrob. Agents Chemother. 2008, 52, 875–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhaldi, A.A.M.; Martinek, J.; Panicucci, B.; Dardonville, C.; Ziková, A.; De Koning, H.P. Trypanocidal action of bisphosphonium salts through a mitochondrial target in bloodstream form Trypanosoma brucei. Int. J. Parasitol. Drugs Drug. Res. 2016, 6, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Ebiloma, G.U.; Balogun, E.O.; Cueto Díaz, E.J.; De Koning, H.P.; Dardonville, C. Alternative Oxidase inhibitors: Development and efficient mitochondrion-targeting as a strategy for new drugs against pathogenic parasites and fungi. Med. Res. Rev. 2019, 39, 1553–1602. [Google Scholar] [CrossRef]
- Fueyo González, F.J.; Ebiloma, G.U.; Izquierdo García, C.; Bruggeman, V.; Sánchez Villamañán, J.M.; Donachie, A.; Balogun, E.O.; Inaoka, D.K.; Shiba, T.; Harada, S.; et al. Conjugates of 2,4-dihydroxybenzoate and salicylhydroxamate and lipocations display potent anti-parasite effects by efficiently targeting the Trypanosoma brucei and Trypanosoma congolense mitochondrion. J. Med. Chem. 2017, 60, 1509–1522. [Google Scholar] [CrossRef] [Green Version]
- Basselin, M.; Denise, H.; Coombs, G.H.; Barrett, M.P. Resistance to pentamidine in Leishmania mexicana involves exclusion of the drug from the mitochondrion. Antimicrob. Agents Chemother. 2002, 46, 3731–3738. [Google Scholar] [CrossRef] [Green Version]
- Bray, P.G.; Barrett, M.P.; Ward, S.A.; De Koning, H.P. Pentamidine uptake and resistance in pathogenic protozoa. Trends Parasitol. 2003, 19, 232–239. [Google Scholar] [CrossRef]
- Doua, F.; Miezan, T.W.; Sanon Singaro, J.R.; Boa Yapo, F.; Baltz, T. The efficacy of pentamidine in the treatment of early-late stage Trypanosoma brucei gambiense trypanosomiasis. Am. J. Trop. Med. Hyg. 1996, 55, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, G.N.; Georgian, A.R.; Sanderson, L.; Vizcay-Barrena, G.; Brown, R.C.; Muresan, P.; Fleck, R.A.; Thomas, S.A. Organic cation transporter 1 (OCT1) is involved in pentamidine transport at the human and mouse blood-brain barrier (BBB). PLoS ONE 2017, 12, e0173474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturk, L.M.; Brock, J.L.; Bagnell, C.R.; Hall, J.E.; Tidwell, R.R. Distribution and quantitation of the anti-trypanosomal diamidine 2,5-bis (4-amidinophenyl) furan (DB75) and its N-methoxy prodrug DB289 in murine brain tissue. Acta Trop. 2004, 91, 131–143. [Google Scholar] [CrossRef]
- Sanderson, L.; Dogruel, M.; Rodgers, J.; De Koning, H.P.; Thomas, S.A. Pentamidine movement across the murine blood-brain and blood-CSF barriers; effect of trypanosome infection, combination therapy, P-glycoprotein and MRP. J. Pharmacol. Exp. Ther. 2009, 329, 967–977. [Google Scholar] [CrossRef]
- Yang, S.; Wenzler, T.; Miller, P.N.; Wu, H.; Boykin, D.W.; Brun, R.; Wang, M.Z. Pharmacokinetic comparison to determine the mechanisms underlying the differential efficacies of cationic diamidines against first- and second-stage human African trypanosomiasis. Antimicrob. Agents Chemother. 2014, 58, 4064–4074. [Google Scholar] [CrossRef] [Green Version]
- Myburgh, E.; Coles, J.A.; Ritchie, R.; Kennedy, P.G.; McLatchie, A.P.; Rodgers, J.; Taylor, M.C.; Barrett, M.P.; Brewer, J.M.; Mottram, J.C. In vivo imaging of trypanosome-brain interactions and development of a rapid screening test for drugs against CNS stage trypanosomiasis. PLoS Negl. Trop. Dis. 2013, 7, e2384. [Google Scholar] [CrossRef]
- Paine, M.; Wang, M.; Boykin, D.; Wilson, W.D.; De Koning, H.P.; Olson, C.; Polig, G.; Burri, C.; Brun, R.; Murilla, G.A.; et al. Diamidines for human African trypanosomiasis. Curr. Opin. Investig. Drugs 2010, 11, 876–883. [Google Scholar]
- Pohlig, G.; Bernhard, S.C.; Blum, J.; Burri, C.; Mpanya, A.; Lubaki, J.P.; Mpoto, A.M.; Munungu, B.F.; N’tombe, P.M.; Deo, G.K.; et al. Efficacy and safety of pafuramidine versus pentamidine maleate for treatment of first stage sleeping sickness in a randomized, comparator-controlled, international phase 3 clinical trial. PLoS Negl. Trop. Dis. 2016, 10, e0004363. [Google Scholar] [CrossRef] [Green Version]
- Wenzler, T.; Boykin, D.W.; Ismail, M.A.; Hall, J.E.; Tidwell, R.R.; Brun, R. New treatment option for second-stage African sleeping sickness: In vitro and in vivo efficacy of aza analogs of DB289. Antimicrob. Agents Chemother. 2009, 53, 4185–4192. [Google Scholar] [CrossRef] [Green Version]
- Thuita, J.K.; Wolf, K.K.; Murilla, G.A.; Bridges, A.S.; Boykin, D.W.; Mutuku, J.N.; Liu, Q.; Jones, S.K.; Gem, C.O.; Ching, S.; et al. Chemotherapy of second stage human African trypanosomiasis: Comparison between the parenteral diamidine DB829 and its oral prodrug DB868 in vervet monkeys. PLoS Negl. Trop. Dis. 2015, 9, e0003409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, N.S.; Berger, B.J.; Fairlamb, A.H. Uptake of diamidine drugs by the P2 nucleoside transporter in melarsen-sensitive and -resistant Trypanosoma brucei brucei. J. Biol. Chem. 1995, 270, 28153–28157. [Google Scholar] [PubMed] [Green Version]
- De Koning, H.P.; Jarvis, S.M. Uptake of pentamidine in Trypanosoma brucei brucei is mediated by the P2 adenosine transporter and at least one novel, unrelated transporter. Acta Trop. 2001, 80, 245–250. [Google Scholar] [CrossRef]
- De Koning, H.P.; Anderson, L.F.; Stewart, M.; Burchmore, R.J.S.; Wallace, L.J.M.; Barrett, M.P. The trypanocide diminazene aceturate is accumulated predominantly through the TbAT1 purine transporter; additional insights in diamidine resistance in African trypanosomes. Antimicrob. Agents Chemother. 2004, 48, 1515–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäser, P.; Sütterlin, C.; Kralli, A.; Kaminsky, R. A nucleoside transporter from Trypanosoma brucei involved in drug resistance. Science 1999, 285, 242–244. [Google Scholar]
- Matovu, E.; Stewart, M.; Geiser, F.; Brun, R.; Mäser, P.; Wallace, L.J.M.; Burchmore, R.J.; Enyaru, J.C.K.; Barrett, M.P.; Kaminsky, R.; et al. The mechanisms of arsenical and diamidine uptake and resistance in Trypanosoma brucei. Eukaryot. Cell 2003, 2, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Graf, F.E.; Ludin, P.; Wenzler, T.; Kaiser, M.; Pyana, P.; Büscher, P.; De Koning, H.P.; Horn, D.; Mäser, P. Aquaporin 2 mutations in Trypanosoma b. gambiense field isolates correlate with decreased susceptibility to pentamidine and melarsoprol. PLoS Negl. Trop. Dis. 2013, 7, e2475. [Google Scholar] [CrossRef] [Green Version]
- Munday, J.C.; Tagoe, D.N.A.; Eze, A.A.; Krezdorn, J.A.; Rojas López, K.E.; Alkhaldi, A.A.M.; McDonald, F.; Still, J.; Alzahrani, K.J.; Settimo, L.; et al. Functional analysis of drug resistance-associated mutations in the Trypanosoma brucei adenosine transporter 1 (TbAT1) and the proposal of a structural model for the protein. Mol. Microbiol. 2015, 96, 887–900. [Google Scholar] [CrossRef]
- De Koning, H.P.; Jarvis, S.M. Adenosine transporters in bloodstream forms of T. b. brucei: Substrate recognition motifs and affinity for trypanocidal drugs. Mol. Pharmacol. 1999, 56, 1162–1170. [Google Scholar] [CrossRef]
- Collar, C.J.; Al-Salabi, M.I.; Stewart, M.L.; Barrett, M.P.; Wilson, W.D.; De Koning, H.P. Predictive computational models of substrate binding by a nucleoside transporter. J. Biol. Chem. 2009, 284, 34028–34035. [Google Scholar] [CrossRef] [Green Version]
- De Koning, H.P. Uptake of pentamidine in Trypanosoma brucei brucei is mediated by three distinct transporters. Implications for crossresistance with arsenicals. Mol. Pharmacol. 2001, 59, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, D.; Gould, M.K.; Nerima, B.; Mäser, P.; Burchmore, R.J.S.; De Koning, H.P. Loss of the High Affinity Pentamidine Transporter is responsible for high levels of cross-resistance between arsenical and diamidine drugs in African trypanosomes. Mol. Pharmacol. 2007, 71, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munday, J.C.; Eze, A.A.; Baker, N.; Glover, L.; Clucas, C.; Aguinaga Andrés, D.; Natto, M.J.; Teka, I.A.; McDonald, J.; Lee, R.S.; et al. Trypanosoma brucei Aquaglyceroporin 2 is a high affinity transporter for pentamidine and melaminophenyl arsenic drugs and is the main genetic determinant of resistance to these drugs. J. Antimicrob. Chemother. 2014, 69, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Alsford, S.; Eckert, S.; Baker, N.; Glover, L.; Sanchez-Flores, A.; Leung, K.F.; Turner, D.J.; Field, M.C.; Berriman, M.; Horn, D. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 2012, 482, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.; Glover, L.; Munday, J.C.; Aguinaga Andrés, D.; Barrett, M.P.; De Koning, H.P.; Horn, D. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc. Natl. Acad. Sci. USA 2012, 109, 10996–11001. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Baker, N.; Rothert, M.; Henke, B.; Jeacock, L.; Horn, D.; Beitz, E. Pentamidine is not a permeant but a nanomolar inhibitor of the Trypanosoma brucei Aquaglyceroporin-2. PLoS Pathog. 2016, 2, e1005436. [Google Scholar] [CrossRef] [Green Version]
- Teka, I.A.; Kazibwe, A.; El-Sabbagh, N.; Al-Salabi, M.I.; Ward, C.P.; Eze, A.A.; Munday, J.C.; Mäser, P.; Matovu, E.; Barrett, M.P.; et al. The diamidine diminazene aceturate is a substrate for the High Affinity Pentamidine Transporter: Implications for the development of high resistance levels. Mol. Pharmacol. 2011, 80, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Langreth, S.G.; Balber, A.E. Protein uptake and digestion in bloodstream and culture forms of Trypanosoma brucei. J. Protozool. 1975, 22, 40–55. [Google Scholar] [CrossRef]
- Field, M.C.; Carrington, M. The trypanosome flagellar pocket. Nat. Rev. Microbiol. 2009, 7, 775–786. [Google Scholar] [CrossRef]
- Munday, J.C.; Settimo, L.; De Koning, H.P. Transport proteins determine drug sensitivity and resistance in a protozoan parasite, Trypanosoma brucei. Front. Pharmacol. 2015, 6, 32. [Google Scholar] [CrossRef]
- Alghamdi, A.; Munday, J.C.; Campagnaro, G.; Eze, A.A.; Svensson, F.; Martin Abril, E.; Milic, P.; Dimitriou, A.; Wielinska, J.; Smart, G.; et al. Pentamidine enters Trypanosoma brucei by passing through the pore of the aquaglyceroporin TbAQP2. 2020; (submitted). [Google Scholar]
- Unciti-Broceta, J.D.; Arias, J.L.; Maceira, J.; Soriano, M.; Ortiz-González, M.; Hernández-Quero, J.; Muñóz-Torres, M.; De Koning, H.P.; Magez, S.; Garcia-Salcedo, J.A. Specific cell targeting therapy bypasses drug resistance mechanisms in African trypanosomiasis. PloS Pathog. 2015, 11, e1004942. [Google Scholar] [CrossRef] [PubMed]
- Livingstone D (1858) Arsenic as a remedy for the tsetse bite. Brit. Med. J. 1858, 1, 360–361.
- Bruce, D. Preliminary Report on the Tsetse Fly Disease or Nagana; Zululand: Harrison and Sons, London, 1895. [Google Scholar]
- The African Trypanosomiases; Mulligan, H.W. (Ed.) George Allen and Unwin Ltd.: London, UK, 1970; pp. 41–88. [Google Scholar]
- Jacobs, W.A.; Heidelberger, M. Aromatic arsenic compounds v. N-substituted glycylarsanilic acids. J. Am. Chem. Soc. 1919, 41, 1809–1821. [Google Scholar] [CrossRef] [Green Version]
- Van Nieuwenhove, S. Present strategies in the treatment of human African trypanosomiasis. In Progress in Human African Trypanosomiasis, Sleeping Sickness; Dumas, M., Bouteille, B., Buguet, A., Eds.; Springer: Paris, France, 1999; pp. 253–281. [Google Scholar]
- Dukes, P. Arsenic and old taxa: Subspeciation and drug sensitivity in Trypanosoma brucei. Trans. R. Soc. Trop. Med. Hyg. 1984, 78, 711–725. [Google Scholar] [CrossRef]
- Fourneau, E.; Tréfouël, J.; Tréfouël, T.; Benoit, G. Sur les isomères de l’acide para-oxy-3-amino-phényl-arsinique et de son dérivé acétylé (stovarsol). Bull. Soc. Chim. Fr. 1927, 41, 499–514. [Google Scholar]
- Apted, F.I.C. Treatment of human trypanosomiasis. In The African Trypanosomiases; Mulligan, H.W., Ed.; George Allen and Unwin Ltd.: London, UK, 1970; pp. 684–710. [Google Scholar]
- Ledentu, G.; Daude, J. Essai du traitement de la trypanosomiase humaine par le 270 Fourneau. Ann. Inst. Pasteur 1926, 40, 830–845. [Google Scholar]
- Laveissière, C.; Penchenier, L. Manuel De Lutte Contre La Maladie Du Sommeil; éditions de l’Institut de recherche pour le développement, coll. Didactiques. IRD Editions: Marseille, France, 2005; Volume 4, p. 273. [Google Scholar]
- Ollivier, G.; Legros, D. Trypanosomiase humaine africaine: Historique de la thérapeutique et de ses échecs. Trop. Med. Int. Health 2001, 6, 855–863. [Google Scholar] [CrossRef] [Green Version]
- Rollo, I.M.; Williamson, J.; Lourie, E.M. Studies on the chemotherapy of melaminyl arsenicals and antimonials in laboratory trypanosome infections. Ann. Trop. Med. Parasitol. 1949, 43, 194–208. [Google Scholar] [CrossRef]
- Van Hoof, L.; Henrard, C.; Peel, E. Pentamidine is the prevention and treatment of trypanosomiasis. Trans. R. Soc. Trop. Med. Hyg. 1944, 37, 271–280. [Google Scholar] [CrossRef]
- Murgatroyd, F.; Yorke, W. Studies in chemotherapy XIV—The stability of drug-resistance in trypanosomes. Ann. Trop. Med. Parsitol. 1937, 31, 165–172. [Google Scholar] [CrossRef]
- Wery, M. therapy for African trypanosomiasis. Curr. Opin. Infect. Dis. 1991, 4, 838–843. [Google Scholar] [CrossRef]
- Jonchere, H.; Gomer, J.; Reynaud, R. Contribution à l’étude de produits a radical mélaminyl dans le traitement de la trypanosmiase humaine à Tr gambiense. Bull. Soc. Pathol. Exot. 1953, 3, 386–396. [Google Scholar]
- Blum, J.; Nkunku, S.; Burri, C. Clinical description of encephalopathic syndromes and risk factors for their occurrence and outcome during melarsoprol treatment of human African trypanosomiasis. Trop. Med. Int. Health 2001, 6, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pépin, J.; Milord, F.; Khonde, A.; Niyonsenga, T.; Loko, L.; Mpia, B. Gambiense trypanosomiasis: Frequency of, and risk factors for, failure of melarsoprol therapy. Trans. R. Soc. Trop. Med. Hyg. 1994, 88, 447–452. [Google Scholar] [CrossRef]
- Schmid, C.; Richer, M.; Bilenge, C.M.; Josenando, T.; Chappuis, F.; Manthelot, C.R.; Nangouma, A.; Doua, F.; Asumu, P.N.; Simarro, P.P.; et al. Effectiveness of a 10-day melarsoprol schedule for the treatment of late-stage human African trypanosomiasis: Confirmation from a multinational study (IMPAMEL II). J. Infect. Dis. 2005, 191, 1922–1931. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.; Jones, A.; Gibaud, S.; Bradley, B.; McCabe, C.; Barrett, M.P.; Gettinby, G.; Kennedy, P.G.E. Melarsoprol Cyclodextrin Inclusion Complexes as Promising Oral Candidates for the Treatment of Human African Trypanosomiasis. PLOS Negl. Trop. Dis. 2011, 5, e1308. [Google Scholar] [CrossRef] [Green Version]
- Committee for Orphan Medicinal Products (COMP) Meeting Report on the Review of Applications for Orphan Designation. 8 October 2012. Available online: https://www.ema.europa.eu/en/documents/committee-report/comp-meeting-report-review-applications-orphan-designation-october-2012_en.pdf (accessed on 17 January 2020).
- Rollo, I.M.; Williamson, J. Acquired resistance to ‘Melarsan’, tryparsamidine and amidines in pathogenic trypanosomes. Nature 1951, 167, 147–148. [Google Scholar] [CrossRef]
- Baker, N.; De Koning, H.P.; Mäser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef] [Green Version]
- De Koning, H.P. The ever-increasing complexities of arsenical-diamidine cross-resistance in African trypanosomes. Trends Parasitol. 2008, 24, 345–349. [Google Scholar] [CrossRef]
- Carter, N.S.; Fairlamb, A.H. Arsenical-resistant trypanosomes lack an unusual adenosine transporter. Nature 1993, 361, 173–176. [Google Scholar] [CrossRef] [PubMed]
- De Koning, H.P.; MacLeod, A.; Barrett, M.P.; Cover, B.; Jarvis, S.M. Further evidence for a link between melarsoprol resistance and P2 transporter function in African trypanosomes. Mol. Biochem. Parasitol. 2000, 106, 181–185. [Google Scholar] [CrossRef]
- Carter, N.S.; Barrett, M.P.; De Koning, H.P. A drug resistance determinant from Trypanosoma brucei. Trends Microbiol. 1999, 7, 469–471. [Google Scholar] [CrossRef]
- Nerima, B.; Matovu, E.; Lubega, G.W.; Enyaru, J.C. Detection of mutant P2 adenosine transporter (TbAT1) gene in Trypanosoma brucei gambiense isolates from northwest Uganda using allele-specific polymerase chain reaction. Trop. Med. Int. Health 2007, 12, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Matovu, E.; Geiser, F.; Schneider, V.; Mäser, P.; Enyaru, J.C.; Kaminsky, R.; Gallati, S.; Seebeck, T. Genetic variants of the TbAT1 adenosine transporter from African trypanosomes in relapse infections following melarsoprol therapy. Mol. Biochem. Parasitol. 2001, 117, 73–81. [Google Scholar] [CrossRef]
- Barrett, M.P.; Vincent, I.M.; Burchmore, R.J.S.; Kazibwe, A.J.N.; Matovu, E. Drug resistance in human African trypanosomiasis. Future Microbiol. 2011, 6, 1037–1047. [Google Scholar] [CrossRef]
- Brun, R.; Schumacher, R.; Schmid, C.; Kunz, C.; Burri, C. The phenomenon of treatment failures in Human African Trypanosomiasis. Trop. Med. Int. Health 2011, 6, 906–914. [Google Scholar] [CrossRef] [Green Version]
- Legros, D.; Fournier, C.; Gastellu Etchegorry, M.; Maiso, F.; Szumilin, E. Therapeutic failure of melarsoprol among patients treated for late stage T.b. gambiense human African trypanosomiasis in Uganda. Bull. Soc. Pathol. Exot. 1999, 92, 171–172. [Google Scholar]
- Moore, A.; Richer, M. Re-emergence of epidemic sleeping sickness in southern Sudan. Trop. Med. Int. Health 2001, 6, 342–347. [Google Scholar] [CrossRef] [Green Version]
- Robays, J.; Nyamowala, G.; Sese, C.; Betu Ku Mesu Kande, V.; Lutumba, P.; Van der Veken, W.; Boelaert, M. High failure rates of melarsoprol for sleeping sickness, Democratic Republic of Congo. Emerg. Infect. Dis. 2008, 14, 966–967. [Google Scholar] [CrossRef]
- Burri, C.; Keiser, J. Pharmacokinetic investigations in patients from northern Angola refractory to melarsoprol treatment. Trop. Med. Int. Health 2001, 6, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Kazibwe, A.J.N.; Nerima, B.; De Koning, H.P.; Mäser, P.; Barrett, M.P.; Matovu, E. Genotypic status of the TbAT1/P2 adenosine transporter of Trypanosoma brucei gambiense isolates from North western Uganda following melarsoprol withdrawal. PLoS Negl. Trop. Dis. 2009, 3, e523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyana Pati, P.; Van Reet, N.; Mumba Ngoyi, D.; Ngay Lukusa, I.; Karhemere Bin Shamamba, S.; Büscher, P. Melarsoprol sensitivity profile of Trypanosoma brucei gambiense isolates from cured and relapsed sleeping sickness patients from the Democratic Republic of the Congo. PLoS Negl. Trop. Dis. 2014, 8, e3212. [Google Scholar] [CrossRef] [Green Version]
- Graf, F.E.; Baker, N.; Munday, J.C.; De Koning, H.P.; Horn, D.; Mäser, P. Chimerization at the AQP2-AQP3 locus is the genetic basis of melarsoprol-pentamidine cross-resistance in clinical Trypanosoma brucei gambiense isolates. Int. J. Parasitol. Drugs Drug Res. 2015, 5, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, F.E.; Ludin, P.; Arquint, C.; Schmidt, R.S.; Schaub, N.; Kunz-Renggli, C.; Munday, J.C.; Krezdorn, J.; Baker, N.; Horn, D.; et al. Comparative genomics of drug resistance of the sleeping sickness parasite Trypanosoma brucei rhodesiense. Cell. Mol. Life. Sci. 2016, 73, 3387–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munday, J.C.; Rojas López, K.E.; Eze, A.A.; Delespaux, V.; Van Den Abbeele, J.; Rowan, T.; Barrett, M.P.; Morrison, L.J.; De Koning, H.P. Functional expression of TcoAT1 reveals it to be a P1-type nucleoside transporter with no capacity for diminazene uptake. Int. J. Parasitol. Drugs Drug Resist. 2013, 3, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, P.; Shiga, K. Farben therapeutische Versuche bei Trypanosomerkrankung. Berl. Klin. Wochenschr. 1904, 14, 362–365. [Google Scholar]
- Ehrlich, P.; Shiga, K. Farbentherapeutische Versuche bei Trypanosomenerkrankung. Berl. Klin. Wochenschr. 1904, 41, 329–332. [Google Scholar]
- Steverding, D. The development of drugs for treatment of sleeping sickness: A historical review. Parasit. Vectors 2010, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, P. Aus Theorie und Praxis der Chemotherapie. Folia Serol. 1911, 7, 697–714. [Google Scholar]
- Nicolle, M.; Mesnil, F. Traitement des trypanosomiases par les couleurs de benzidine. Première partie—Étude chemique. Ann. Inst. Pasteur. 1906, 20, 417–448. [Google Scholar]
- Mesnil, F.; Nicolle, M. Traitement des trypanosomiases par les couleurs de benzidine. Second partie—Étude expérimentale. Ann. Inst. Pasteur. 1906, 20, 513–538. [Google Scholar]
- Tréfouël, J. Le rôle de Maurice Nicolle en chimiothérapie anti-trypanosome. Bull. Soc. Pathol. Exotique. 1962, 55, 200–207. [Google Scholar]
- Travis, A.S. Paul Ehrlich: A hundred years of chemotherapy 1891-1991. Biochemist 1991, 13, 9–12. [Google Scholar]
- Schlitzer, M. Wirkstoffe zur Behandlung der Afrikanischer Schlafkrankheit. Pharm. Unsere Zeit. 2009, 6, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.J. Synthetic therapeutic agents. Br. Med. J. 1924, 1, 413–414. [Google Scholar] [CrossRef] [Green Version]
- Fourneau, E.; Tréfouël, J.; Vallée, J. Sur une nouvelle série de médicaments trypanocides. Comptes Rendus des Séances de l’Académie des Sciences 1924, 178, 675. [Google Scholar]
- Fourneau, E.; Tréfouël, J.; Vallée, J. Recherches de chimiothérapie dans la série du 205 Bayer. Urées des acides aminobenzoylaminonaphtaléniques. Ann. Inst. Pasteur. 1924, 38, 81–114. [Google Scholar]
- Apted, F.I.C. Present status of chemotherapy and chemoprophylaxis of human trypanosomiasis in the Eastern hemisphere. Pharmac. Ther. 1980, 11, 391–413. [Google Scholar] [CrossRef]
- Pépin, J.; Milord, F. The treatment of human African trypanosomiasis. Adv. Parasitol. 1994, 33, 1–47. [Google Scholar]
- Scott, A.G.; Tait, A.; Turner, C.M. Characterisation of cloned lines of Trypanosoma brucei expressing stable resistance to MelCy and suramin. Acta Trop. 1996, 60, 251–262. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Coppens, I.; Opperdoes, F.R.; Courtoy, P.J.; Baudhuin, P. Receptor-mediated endocytosis in the blood-stream form of Trypanosoma brucei. J. Protozool. 1987, 34, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Vansterkenburg, E.L.M.; Coppens, I.; Wilting, J.; Bos, O.J.M.; Fischer, M.J.E.; Janssen, L.H.M.; Opperdoes, F.R. The uptake of the trypanocidal drug Suramin in combination with low-density lipoproteins by Trypanosoma brucei and its possible mode of action. Acta Trop. 1993, 54, 237–250. [Google Scholar] [CrossRef]
- Pal, A.; Hall, B.S.; Field, M.C. Evidence for a non-LDL-mediated entry route for the trypanocidal drug suramin in Trypanosoma brucei. Mol. Biochem. Parasitol. 2002, 122, 217–221. [Google Scholar] [CrossRef]
- Jaffe, J.J.; McCormack, J.J.; Meymariam, E. Comparative properties of schistosomal and filarial dihydrofolate reductase. Biochem. Pharmacol. 1972, 21, 719–731. [Google Scholar] [CrossRef]
- Chello, P.L.; Jaffe, J.J. Comparative properties of trypanosomal and mammalian thymidine kinases. Comp. Biochem. Physiol. B 1972, 43, 543–562. [Google Scholar] [CrossRef]
- Willson, M.; Callens, M.; Kuntz, D.A.; Perié, J.; Opperdoes, F.R. Synthesis and activity of inhibitors highly specific for the glycolytic enzymes from Trypanosoma brucei. Mol. Biochem. Parasitol. 1993, 59, 201–210. [Google Scholar] [CrossRef]
- Zoltner, M.; Horn, D.; De Koning, H.P.; Field, M.C. Exploiting the Achilles’ heel of membrane trafficking in trypanosomes. Curr. Opin. Microbiol. 2016, 34, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Alsford, S.; Field, M.C.; Horn, D. Receptor-mediated endocytosis for drug delivery in African trypanosomes: Fulfilling Paul Ehrlich’s vision of chemotherapy. Trends. Parasitol. 2013, 29, 207–212. [Google Scholar] [CrossRef]
- Poulin, R.; Lu, L.; Ackermann, B.; Bey, P.; Pegg, A.E. Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by alpha-difluoromethylornithine. Characterization of sequences at the inhibitor and coenzyme binding sites. J. Biol. Chem. 1992, 267, 150–158. [Google Scholar]
- Weeks, C.E.; Herrmann, A.L.; Nelson, F.R.; Slaga, T.J. α-Difluoromethylornithine, an irreversible inhibitor of ornithine decarboxylase, inhibits tumor promoter-induced polyamine accumulation and carcinogenesis in mouse skin. Proc. Natl. Acad. Sci. USA 1982, 79, 6028–6032. [Google Scholar] [CrossRef] [Green Version]
- Meyskens, F.L., Jr.; Gerner, E.W. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. 1999, 5, 945–951. [Google Scholar] [PubMed]
- Gerner, E.W.; Meyskens, F.L., Jr. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792. [Google Scholar] [CrossRef] [Green Version]
- Milord, F.; Pépin, J.; Loko, L.; Ethier, L.; Mpia, B. Efficacy and toxicity of eflornithine for treatment of Trypanosoma brucei gambiense sleeping sickness. Lancet 1992, 340, 652–655. [Google Scholar] [CrossRef]
- Van Nieuwenhove, S.; Schechter, P.J.; Declercq, J.; Bone, G.; Burke, J.; Sjoerdsma, A. Treatment of gambiense sleeping sickness in Sudan with oral DFMO (DL-alpha-difluoromethylornithine), an inhibitor of ornithine decarboxylase: First field trial. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 692–698. [Google Scholar] [CrossRef]
- Taelman, H.; Schechter, P.J.; Marcelis, L.; Sonnet, J.; Kazyumba, G.; Dasnoy, J.; Haegele, K.D.; Sjoerdsma, A.; Wery, M. Difluoromethylornithine, an effective new treatment of Gambian trypanosomiasis. Results in five patients. Am. J. Med. 1987, 82, 607–614. [Google Scholar] [CrossRef]
- Eozenou, P.; Jannin, J.; Ngampo, S.; Carme, B.; Tell, G.P.; Schechter, P.J. Essai de traitement de la trypanosomiase a Trypanosoma brucei gambiense par l’eflornithine en Republique Populaire du Congo. Med. Trop. 1989, 49, 149–154. [Google Scholar]
- Ebikeme, C. The death and life of the resurrection drug. PLoS Negl. Trop. Dis. 2014, 8, e2910. [Google Scholar] [CrossRef] [Green Version]
- Burri, C.; Brun, R. Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res. 2003, 90, S49–S52. [Google Scholar] [CrossRef]
- Kennedy, P.G. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013, 12, 186–194. [Google Scholar] [CrossRef]
- Bales, J.D.; Harison, S.M.; Mbwabi, D.L.; Schechter, P.J. Treatment of arsenical refractory Rhodesian sleeping sickness in Kenya. Ann. Trop. Med. Parasitol. 1989, 83 (Suppl. S1), 111–114. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, C.J.; Nathan, H.C.; Livingston, T.; Valladares, G.; Saric, M.; Sayer, P.D.; Njogu, A.R.; Clarkson, A.B., Jr. Differential susceptibility to DL-alpha-difluoromethylornithine in clinical isolates of Trypanosoma brucei rhodesiense. Antimicrob. Agents Chemother. 1990, 34, 1183–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iten, M.; Matovu, E.; Brun, R.; Kaminsky, R. Innate lack of susceptibility of Ugandan Trypanosoma brucei rhodesiense to DL-alpha-difluoromethylornithine (DFMO). Trop. Med. Parasitol. 1995, 46, 190–194. [Google Scholar] [PubMed]
- Iten, M.; Mett, H.; Evans, A.; Enyaru, J.C.; Brun, R.; Kaminsky, R. Alterations in ornithine decarboxylase characteristics account for tolerance of Trypanosoma brucei rhodesiense to d,l-alpha-difluoromethylornithine. Antimicrob. Agents Chemother. 1997, 41, 1922–1925. [Google Scholar] [CrossRef] [Green Version]
- Bellofatto, V.; Fairlamb, A.H.; Henderson, G.B.; Cross, G.A.M. Biochemical changes associated with alpha-difluoromethylornithine uptake and resistance in Trypanosoma brucei. Mol. Biochem. Parasitol. 1987, 25, 227–238. [Google Scholar] [CrossRef]
- Phillips, M.A.; Wang, C.C. A Trypanosoma brucei mutant resistant to alpha-difluoromethylornithine. Mol. Biochem. Parasitol. 1987, 22, 9–17. [Google Scholar] [CrossRef]
- Delespaux, V.; De Koning, H.P. Drugs and drug resistance in African trypanosomiasis. Drug Resist. Updat. 2007, 10, 30–50. [Google Scholar] [CrossRef]
- Bitonti, A.J.; Bacchi, C.J.; McCann, P.P.; Sjoerdsma, A. Uptake of alpha-difluoromethylornithine by Trypanosoma brucei brucei. Biochem. Pharmacol. 1986, 35, 351–354. [Google Scholar] [CrossRef]
- Vincent, I.M.; Creek, D.; Watson, D.G.; Kamleh, M.A.; Woods, D.J.; Wong, P.E.; Burchmore, R.J.; Barrett, M.P. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. 2010, 6, e1001204. [Google Scholar] [CrossRef] [Green Version]
- Schumann Burkard, G.; Jutzi, P.; Roditi, I. Genome-wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol. Biochem. Parasitol. 2011, 175, 91–94. [Google Scholar] [CrossRef]
- Simarro, P.P.; Franco, J.; Diarra, A.; Postigo, J.A.; Jannin, J. Update on field use of the available drugs for the chemotherapy of human African trypanosomiasis. Parasitology 2012, 139, 842–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mpia, B.; Pépin, J. Combination of eflornithine and melarsoprol for melarsoprol-resistant Gambian trypanosomiasis. Trop. Med. Int. Health 2002, 7, 775–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priotto, G.; Fogg, C.; Balasegaram, M.; Erphas, O.; Louga, A.; Checchi, F.; Ghabri, S.; Piola, P. Three drug combinations for late-stage Trypanosoma brucei gambiense sleeping sickness: A randomized clinical trial in Uganda. PLoS Clin. Trials. 2006, 1, e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checchi, F.; Piola, P.; Ayikoru, H.; Thomas, F.; Legros, D.; Priotto, G. Nifurtimox plus Eflornithine for late-stage sleeping sickness in Uganda: A case series. PLoS Negl. Trop. Dis. 2007, 1, e64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priotto, G.; Kasparian, S.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Karunakara, U. Nifurtimox-eflornithine combination therapy for second-stage Trypanosoma brucei gambiense sleeping sickness: A randomized clinical trial in Congo. Clin. Infect. Dis. 2007, 45, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S.; et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, randomised, phase III, non-inferiority trial. Lancet 2009, 374, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Yun, O.; Priotto, G.; Tong, J.; Flevaud, L.; Chappuis, F. NECT is next: Implementing the new drug combination therapy for Trypanosoma brucei gambiense sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e720. [Google Scholar] [CrossRef] [Green Version]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Ruiz-Postigo, J.A.; Samo, M.; Jannin, J.G. Monitoring the use of nifurtimox-eflornithine combination therapy (NECT) in the treatment of second stage gambiense human African trypanosomiasis. Res. Rep. Trop. Med. 2012, 3, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Alirol, E.; Schrumpf, D.; Amici Heradi, J.; Riedel, A.; de Patoul, C.; Quere, M.; Chappuis, F. Nifurtimox-eflornithine combination therapy for second-stage gambiense human African trypanosomiasis: Médecins Sans Frontières experience in the Democratic Republic of the Congo. Clin. Infect. Dis. 2013, 56, 195–203. [Google Scholar] [CrossRef]
- Kansiime, F.; Adibaku, S.; Wamboga, C.; Idi, F.; Kato, C.D.; Yamuah, L.; Vaillant, M.; Kioy, D.; Olliaro, P.; Matovu, E. A multicentre, randomised, non-inferiority clinical trial comparing a nifurtimox-eflornithine combination to standard eflornithine monotherapy for late stage Trypanosoma brucei gambiense human African trypanosomiasis in Uganda. Parasit. Vectors 2018, 11, 105. [Google Scholar] [CrossRef]
- Wegner, D.H.G.; Rohwedder, R.W. The effect of nifurtimox on acute Chagas’ infection. Arzneim. Forsch. 1972, 22, 1624–1635. [Google Scholar]
- Ribeiro, V.; Dias, N.; Paiva, T.; Hagström-Bex, L.; Nitz, N.; Pratesi, R.; Hecht, M. Current trends in the pharmacological management of Chagas disease. Int. J. Parasitol. Drugs Drug Resist. 2019, 12, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Janssens, P.G.; De Muynck, A. Clinical trials with “nifurtimox” in African trypanosomiasis. Ann. Soc. Belg. Med. Trop. 1977, 57, 475–479. [Google Scholar] [PubMed]
- Moens, F.; De Wilde, M.; Ngato, K. Essai de traitement au nifurtimox de la trypanosomiase humaine Africaine. Ann. Soc. Belg. Med. Trop. 1984, 64, 37–43. [Google Scholar]
- Pepin, J.; Milord, F.; Mpia, B.; Meurice, F.; Ethier, L.; DeGroof, D.; Bruneel, H. An open clinical trial of nifurtimox for arseno-resistant Trypanosoma brucei gambiense sleeping sickness in central Zaire. Trans. R. Soc. Trop. Med. Hyg. 1989, 83, 514–517. [Google Scholar] [CrossRef]
- Pépin, J.; Milord, F.; Meurice, F.; Ethier, L.; Loko, L.; Mpia, B. High-dose nifurtimox for arseno-resistant Trypanosoma brucei gambiense sleeping sickness: An open trial in central Zaire. Trans. R. Soc. Trop. Med. Hyg. 1992, 86, 254–256. [Google Scholar] [CrossRef]
- Van Nieuwenhove, S. Advances in sleeping sickness therapy. Ann. Soc. Belg. Med. Trop. 1992, 72 (Suppl. S1), 39–51. [Google Scholar]
- Bisser, S.; N’Siesi, F.X.; Lejon, V.; Preux, P.M.; Van Nieuwenhove, S.; Miaka Mia Bilenge, C.; Būscher, P. Equivalence trial of melarsoprol and nifurtimox monotherapy and combination therapy for the treatment of second-stage Trypanosoma brucei gambiense sleeping sickness. J. Infect. Dis. 2007, 195, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Docampo, R.; Stoppani, A.O. generation of superoxide anion and hydrogen peroxide induced by nifurtimox in Trypanosoma cruzi. Arch. Biochem. Biophys. 1979, 197, 317–321. [Google Scholar] [CrossRef]
- Prathalingham, S.R.; Wilkinson, S.R.; Horn, D.; Kelly, J.M. Deletion of the Trypanosoma brucei superoxide dismutase gene sodb1 increases sensitivity to nifurtimox and benznidazole. Antimicrob. Agents Chemother. 2007, 51, 755–758. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S.R.; Taylor, M.C.; Horn, D.; Kelly, J.M.; Cheeseman, I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 5022–5027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, N.; Alsford, S.; Horn, D. Genome-wide RNAi screens in African trypanosomes identify the nifurtimox activator NTR and the eflornithine transporter AAT6. Mol. Biochem. Parasitol. 2011, 176, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.Y.; Wyllie, S.; Patterson, S.; Oza, S.L.; Read, K.D.; Fairlamb, A.H. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 2010, 54, 2893–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeganathan, S.; Sanderson, L.; Dogruel, M.; Rodgers, J.; Croft, S.; Thomas, S.A. The distribution of nifurtimox across the healthy and trypanosome-infected murine blood–brain and blood-CSF barriers. J. Pharmacol. Exp. Ther. 2010, 336, 506–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, C.P.; Dogruel, M.; Mihoreanu, L.; Begley, D.J.; Weksler, B.B.; Couraud, P.O.; Romero, I.A.; Thomas, S.A. The transport of nifurtimox, an anti-trypanosomal drug, in an in vitro model of the human blood-brain barrier: Evidence for involvement of breast cancer resistance protein. Brain Res. 2012, 1436, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duhm, B.; Maul, W.; Medenwald, H.; Patzschke, K.; Wegner, L.A. Investigations on the pharmacokinetics of nifurtimox-35S in the rat and dog. Arzneimittelforschung 1972, 22, 1617–1624. [Google Scholar] [PubMed]
- Kell, D.B.; Oliver, S.G. How drugs get into cells: Tested and testable predictions to help discriminate between transporter-mediated uptake and lipoidal bilayer diffusion. Front. Pharmacol. 2014, 5, 231. [Google Scholar] [CrossRef] [Green Version]
- Kell, D.B.; Dobson, P.D.; Oliver, S.G. Pharmaceutical drug transport: The issues and the implications that it is essentially carrier-mediated only. Drug Discov. Today 2011, 16, 704–714. [Google Scholar] [CrossRef]
- Baliani, A.; Bueno, G.J.; Stewart, M.L.; Yardley, V.; Brun, R.; Barrett, M.P.; Gilbert, I.H. Design and synthesis of a series of melamine-based nitroheterocycles with activity against Trypanosomatid parasites. J. Med. Chem. 2005, 48, 5570–5579. [Google Scholar] [CrossRef]
- Maina, N.; Maina, K.J.; Mäser, P.; Brun, R. Genotypic and phenotypic characterization of Trypanosoma brucei gambiense isolates from Ibba, South Sudan, an area of high melarsoprol treatment failure rate. Acta Trop. 2007, 104, 84–90. [Google Scholar] [CrossRef]
- Deeks, E.D. Fexinidazole: First global approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef]
- Mesu, V.K.B.K.; Kalonji, W.M.; Bardonneau, C.; Mordt, O.V.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Kuziena, W.; Lubaki, J.F.; et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: A pivotal multicentre, randomised, non-inferiority trial. Lancet 2018, 391, 144–154. [Google Scholar] [CrossRef]
- Pelfrene, E.; Harvey Allchurch, M.; Ntamabyaliro, N.; Nambasa, V.; Ventura, F.V.; Nagercoil, N.; Cavaleri, M. The European Medicines Agency’s scientific opinion on oral fexinidazole for human African trypanosomiasis. PLoS Negl. Trop. Dis. 2019, 13, e0007381. [Google Scholar]
- Chappuis, F. Oral fexinidazole for human African trypanosomiasis. Lancet 2018, 391, 100–102. [Google Scholar] [CrossRef]
- Lindner, A.K.; Lejon, V.; Chappuis, F.; Seixas, J.; Kazumba, L.; Barrett, M.P.; Mwamba, E.; Erphas, O.; Akl, E.A.; Villanueva, G.; et al. New WHO guidelines for treatment of gambiense human African trypanosomiasis including fexinidazole: Substantial changes for clinical practice. Lancet Infect. Dis. 2019. [Google Scholar] [CrossRef]
- De Koning, H.P. Drug resistance in protozoan parasites. Emerg. Top. Life Sci. 2017, 1, 627–632. [Google Scholar]
- Blum, J.; Burri, C. Treatment of late stage sleeping sickness caused by T. b. gambiense: A new approach to the use of an old drug. Swiss. Med. Wkly. 2002, 132, 51–56. [Google Scholar] [PubMed]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
P. De Koning, H. The Drugs of Sleeping Sickness: Their Mechanisms of Action and Resistance, and a Brief History. Trop. Med. Infect. Dis. 2020, 5, 14. https://doi.org/10.3390/tropicalmed5010014
P. De Koning H. The Drugs of Sleeping Sickness: Their Mechanisms of Action and Resistance, and a Brief History. Tropical Medicine and Infectious Disease. 2020; 5(1):14. https://doi.org/10.3390/tropicalmed5010014
Chicago/Turabian StyleP. De Koning, Harry. 2020. "The Drugs of Sleeping Sickness: Their Mechanisms of Action and Resistance, and a Brief History" Tropical Medicine and Infectious Disease 5, no. 1: 14. https://doi.org/10.3390/tropicalmed5010014
APA StyleP. De Koning, H. (2020). The Drugs of Sleeping Sickness: Their Mechanisms of Action and Resistance, and a Brief History. Tropical Medicine and Infectious Disease, 5(1), 14. https://doi.org/10.3390/tropicalmed5010014