

Investigating the Leishmania donovani sacp Gene and Its Role in Macrophage Infection and Survival in Mice
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Leishmania Strain and Culture Media
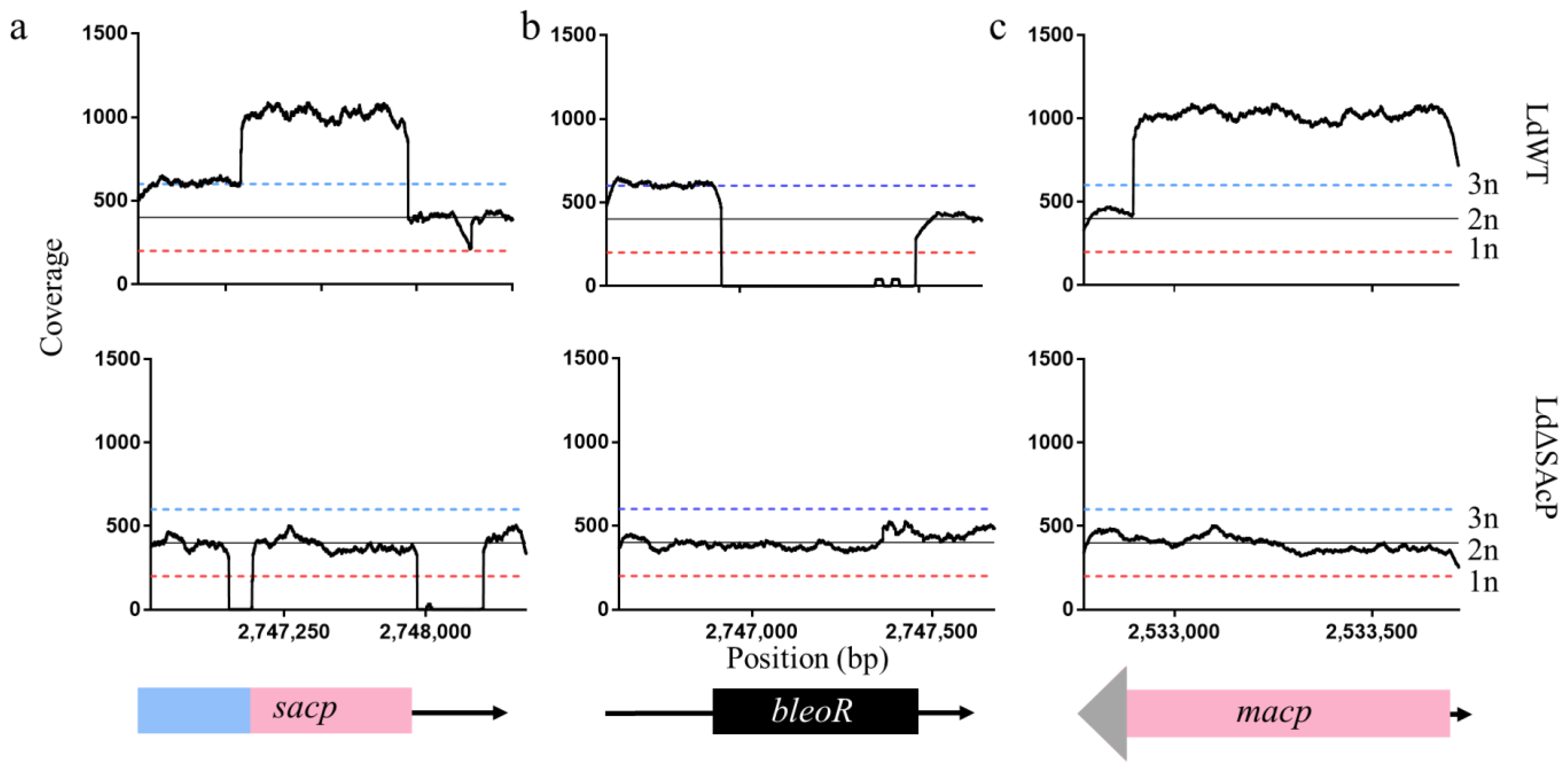
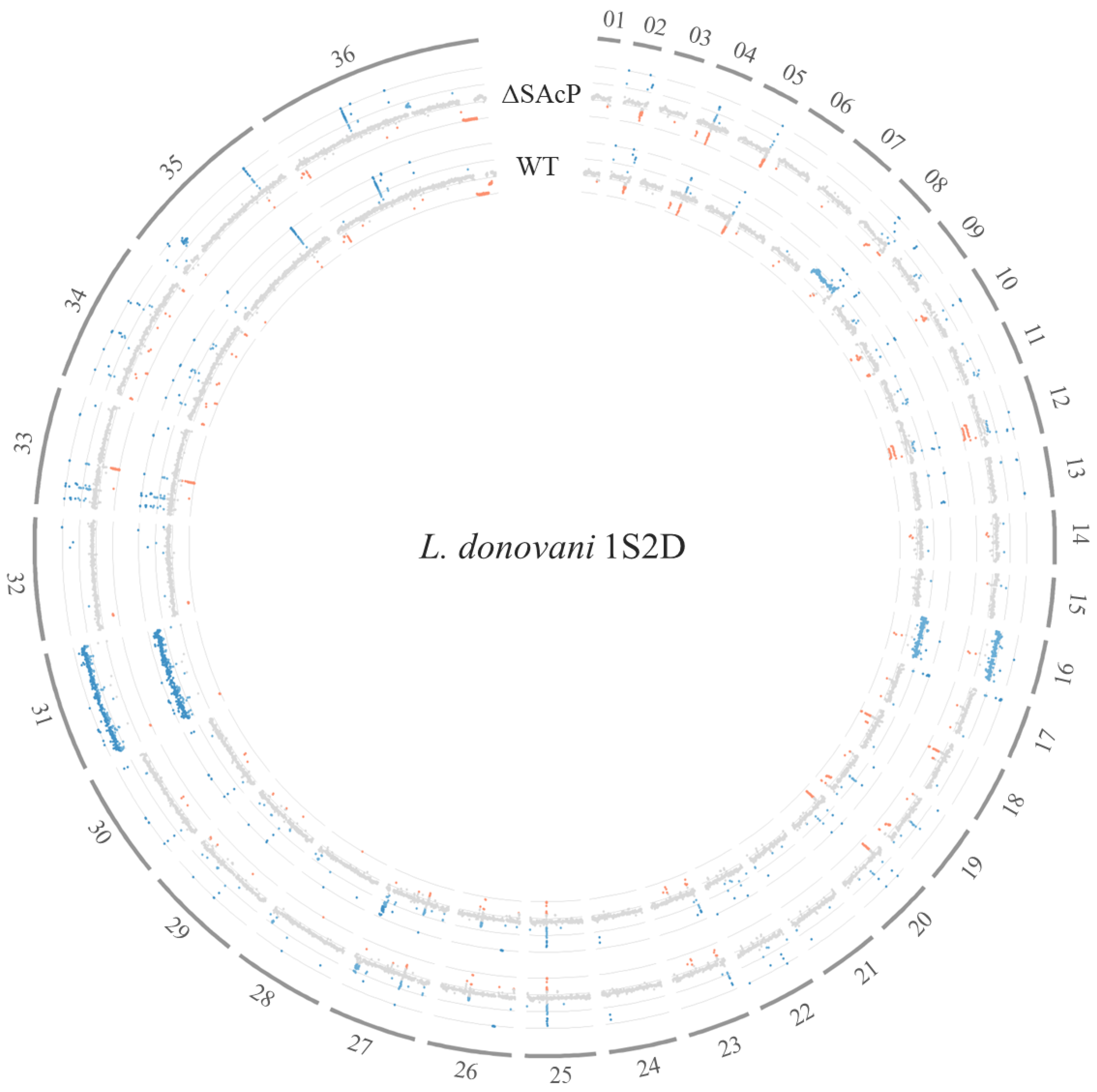
2.2. Whole-Genome Sequencing and Analysis
2.3. Generation and Confirmation of CRISPR-Cas9 Knockouts
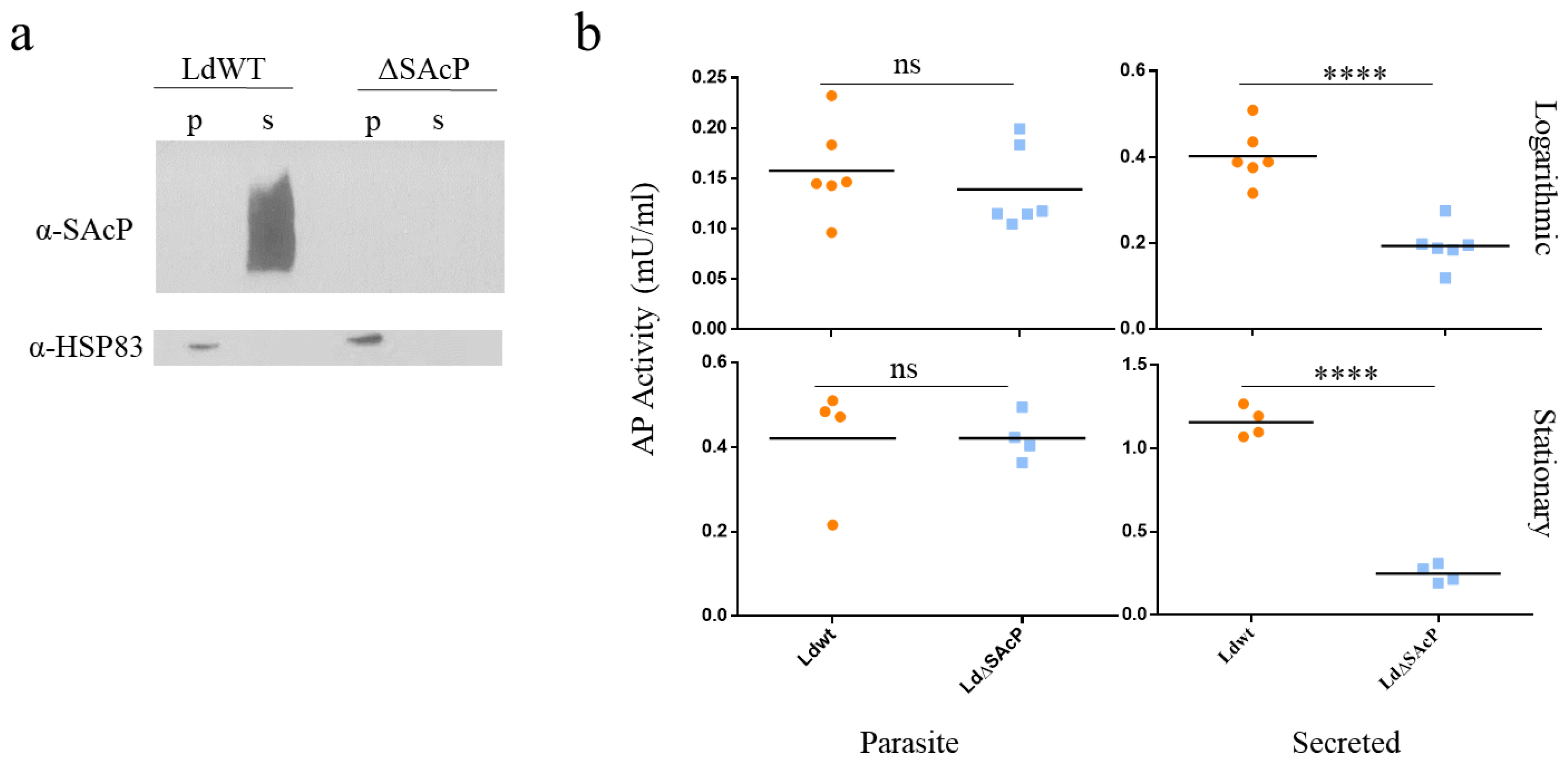
2.4. SDS PAGE and Western Blot Analysis
2.5. Acid Phosphatase Activity Assay
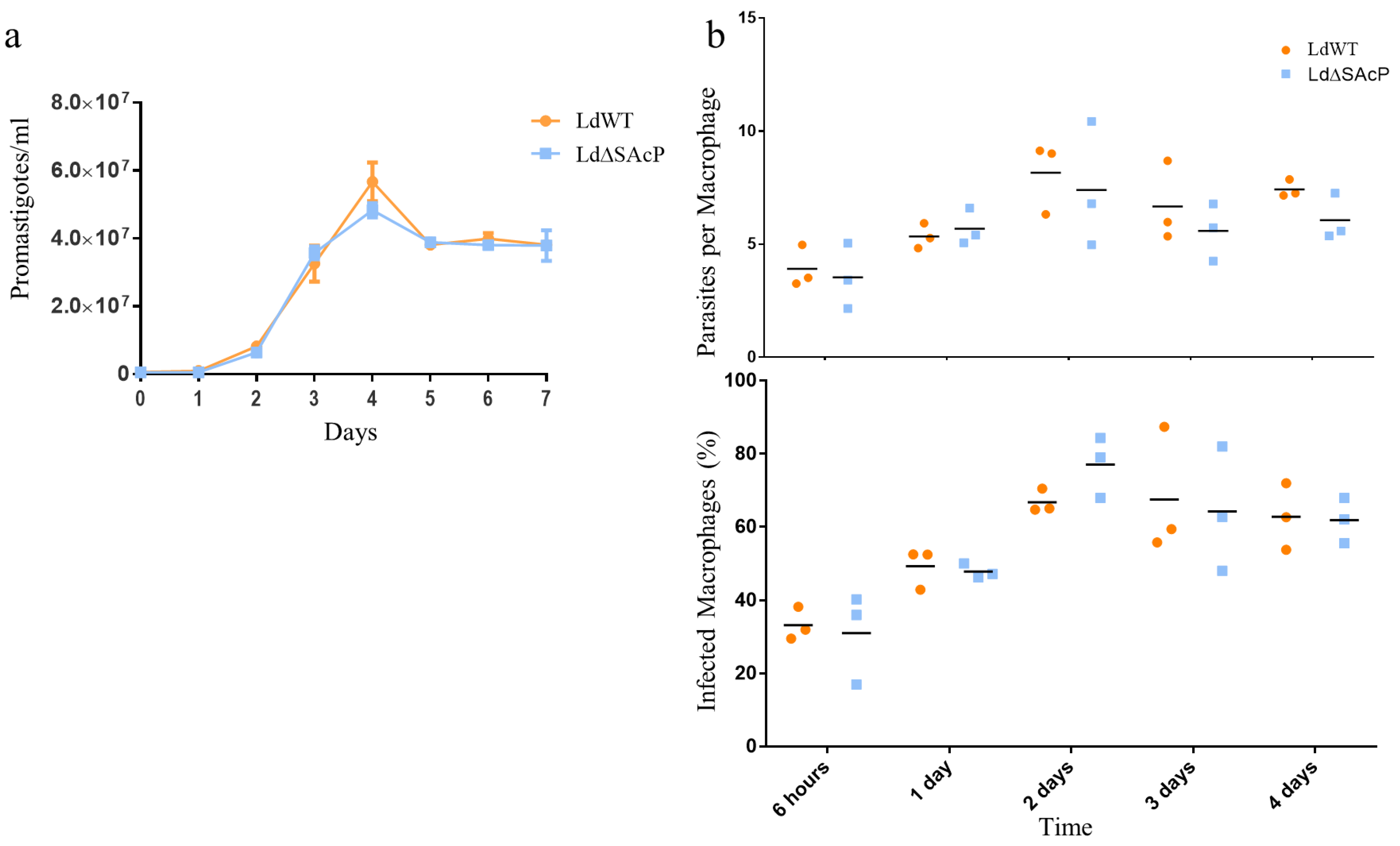
2.6. Promastigote Proliferation Curves
2.7. In Vitro Macrophage Infections
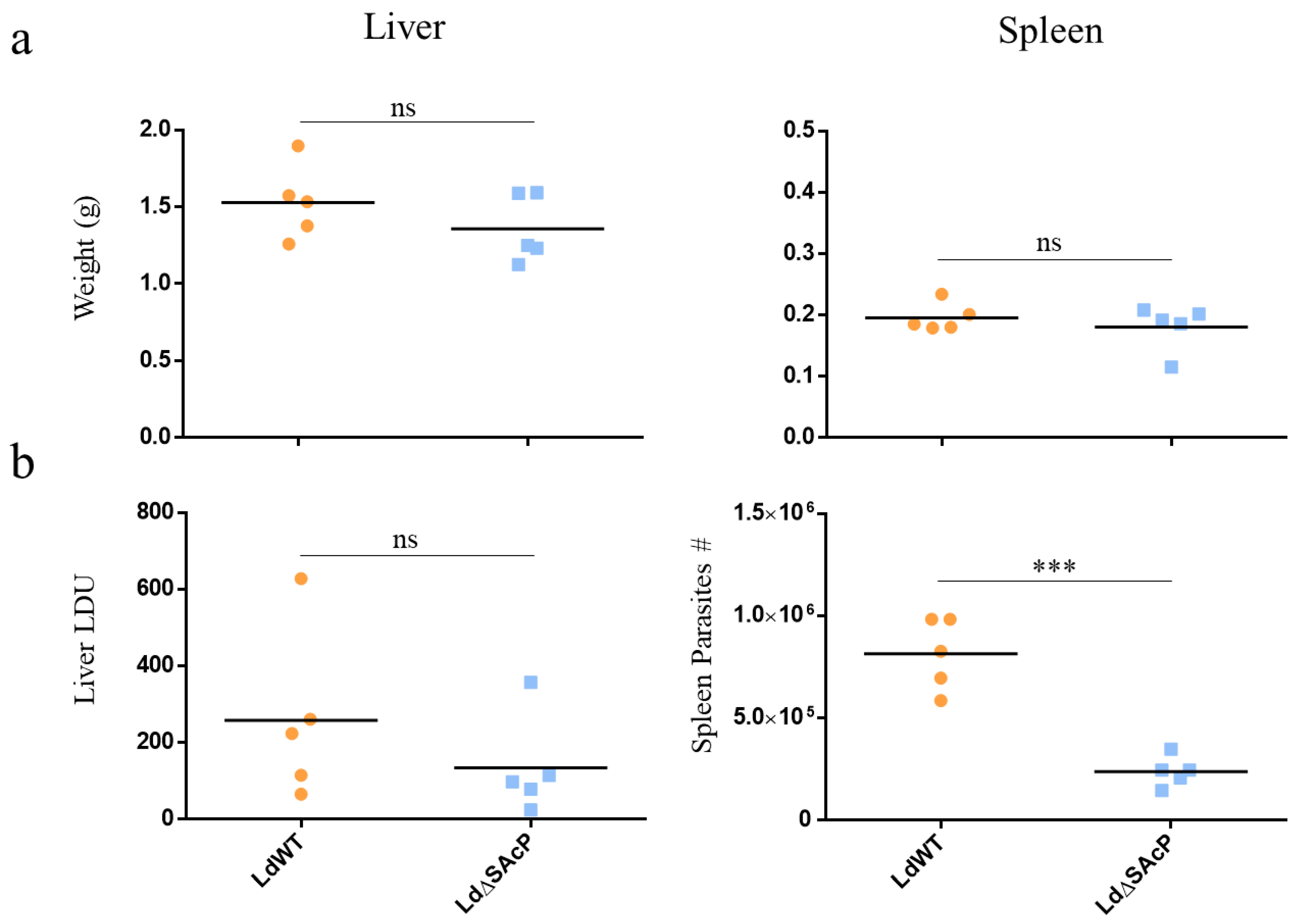
2.8. BALB/c Mouse Infections
3. Results
3.1. sacp Gene Copy Number in the L. donovani 1S2D Used in This Study
3.2. Generation of the L. donovani Mutant, LdΔSAcP, Using CRISPR-Cas9 Gene Editing
3.3. Whole-Genome Sequencing Shows Specificity of CRISPR-Cas9 Gene Knockout
3.4. Comparison of LdWT and LdΔSAcP Promastigotes in Culture and Amastigotes in Macrophages and BALB/c Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- CDC. For D.C. and CDC–Leishmaniasis. Epidemiology & Risk Factors. Available online: https://www.cdc.gov/parasites/leishmaniasis/epi.html (accessed on 26 January 2022).
- CDC. For D.C. and CDC-Leishmaniasis. Biology. Available online: https://www.cdc.gov/parasites/leishmaniasis/biology.html (accessed on 26 January 2022).
- Volpedo, G.; Huston, R.H.; Holcomb, E.A.; Pacheco-Fernandez, T.; Gannavaram, S.; Bhattacharya, P.; Nakhasi, H.L.; Satoskar, A.R. From Infection to Vaccination: Reviewing the Global Burden, History of Vaccine Development, and Recurring Challenges in Global Leishmaniasis Protection. Expert Rev. Vaccines 2021, 20, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Mahajan, S.; Kour, P.; Singh, K. Virulence Factors of Leishmania Parasite: Their Paramount Importance in Unraveling Novel Vaccine Candidates and Therapeutic Targets. Life Sci. 2022, 306, 120829. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Atayde, V.D.; Isnard, A.; Hassani, K.; Shio, M.T. Leishmania Virulence Factors: Focus on the Metalloprotease GP63. Microbes Infect. 2012, 14, 1377–1389. [Google Scholar] [CrossRef]
- McCall, L.-I.; Zhang, W.-W.; Matlashewski, G. Determinants for the Development of Visceral Leishmaniasis Disease. PLoS Pathog. 2013, 9, e1003053. [Google Scholar] [CrossRef] [Green Version]
- Shakarian, A.M.; Joshi, M.B.; Yamage, M.; Ellis, S.L.; Debrabant, A.; Dwyer, D.M. Members of a Unique Histidine Acid Phosphatase Family Are Conserved amongst a Group of Primitive Eukaryotic Human Pathogens. Mol. Cell. Biochem. 2003, 245, 31–41. [Google Scholar] [CrossRef]
- Shakarian, A.M.; Dwyer, D.M. Structurally Conserved Soluble Acid Phosphatases Are Synthesized and Released by Leishmania Major Promastigotes. Exp. Parasitol. 2000, 95, 79–84. [Google Scholar] [CrossRef]
- Bates, P.A.; Kurtz, M.K.; Gottlieb, M.; Dwyer, D.M. Leishmania Donovani: Generation of Monospecific Antibody Reagents to Soluble Acid Phosphatase. Exp. Parasitol. 1987, 64, 157–164. [Google Scholar] [CrossRef]
- Bates, P.A.; Dwyer, D.M. Biosynthesis and Secretion of Acid Phosphatase by Leishmania Donovani Promastigotes. Mol. Biochem. Parasitol. 1987, 26, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb’, M.; Dwyer, D.M. Identification and Partial Characterization of an Extracellular Acid Phosphatase Activity of Leishmania Donovani Promastigotes. Mol. Cell. Biol. 1982, 2, 6. [Google Scholar] [CrossRef]
- Downing, T.; Imamura, H.; Decuypere, S.; Clark, T.G.; Coombs, G.H.; Cotton, J.A.; Hilley, J.D.; de Doncker, S.; Maes, I.; Mottram, J.C.; et al. Whole Genome Sequencing of Multiple Leishmania Donovani Clinical Isolates Provides Insights into Population Structure and Mechanisms of Drug Resistance. Genome Res. 2011, 21, 2143–2156. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.; Catta-Preta, C.M.C.; Neish, R.; Sadlova, J.; Powell, B.; Alves-Ferreira, E.V.C.; Geoghegan, V.; Carnielli, J.B.T.; Newling, K.; Hughes, C.; et al. Systematic functional analysis of Leishmania protein kinases identifies regulators of differentiation or survival. Nat. Commun. 2021, 12, 1244. [Google Scholar] [CrossRef]
- Wiese, M. A Mitogen-Activated Protein (MAP) Kinase Homologue of Leishmania Mexicana Is Essential for Parasite Survival in the Infected Host. EMBO J. 1998, 17, 2619–2628. [Google Scholar] [CrossRef] [Green Version]
- Wiese, M.; Ilg, T.; Lottspeich, F.; Overath, P. Ser/Thr-Rich Repetitive Motifs as Targets for Phosphoglycan Modifications in Leishmania Mexicana Secreted Acid Phosphatase. EMBO J. 1995, 14, 1067–1074. [Google Scholar] [CrossRef]
- Remaley, A.T.; Glew, R.H.; Kuhns, D.B.; Basford, R.E.; Waggoner, A.S.; Ernst, L.A.; Pope, M. Leishmania Donovani: Surface Membrane Acid Phosphatase Blocks Neutrophil Oxidative Metabolite Production. Exp. Parasitol. 1985, 60, 331–341. [Google Scholar] [CrossRef]
- Saha, A.K.; Das, S.; Glew, R.H.; Gottlieb, M. Resistance of Leishmanial Phosphatases to Inactivation by Oxygen Metabolites. J. Clin. Microbiol. 1985, 22, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Remaley, A.T.; Kuhns, D.B.; Basford, R.E.; Glew, R.H.; Kaplan, S.S. Leishmanial Phosphatase Blocks Neutrophil O-2 Production. J. Biol. Chem. 1984, 259, 11173–11175. [Google Scholar] [CrossRef]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas Guides the Future of Genetic Engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-W.; Matlashewski, G. CRISPR-Cas9-Mediated Genome Editing in Leishmania Donovani. mBio 2015, 6, e00861-15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-W.; Lypaczewski, P.; Matlashewski, G. Optimized CRISPR-Cas9 Genome Editing for Leishmania and Its Use To Target a Multigene Family, Induce Chromosomal Translocation, and Study DNA Break Repair Mechanisms. mSphere 2017, 2, e00340-16. [Google Scholar] [CrossRef]
- Lypaczewski, P.; Hoshizaki, J.; Zhang, W.-W.; McCall, L.-I.; Torcivia-Rodriguez, J.; Simonyan, V.; Kaur, A.; Dewar, K.; Matlashewski, G. A Complete Leishmania Donovani Reference Genome Identifies Novel Genetic Variations Associated with Virulence. Sci. Rep. 2018, 8, 16549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinforma. Oxf. Engl. 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- The Galaxy Community. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2022 Update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-W.; Lypaczewski, P.; Matlashewski, G. Application of CRISPR/Cas9-Mediated Genome Editing in Leishmania. In Trypanosomatids; Michels, P.A.M., Ginger, M.L., Zilberstein, D., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; Volume 2116, pp. 199–224. ISBN 978-1-07-160293-5. [Google Scholar]
- Peng, D.; Tarleton, R. EuPaGDT: A Web Tool Tailored to Design CRISPR Guide RNAs for Eukaryotic Pathogens. Microb. Genom. 2015, 1, e000033. [Google Scholar] [CrossRef]
- Zhang, W.-W.; Matlashewski, G. Screening Leishmania Donovani Complex-Specific Genes Required for Visceral Disease. In Parasite Genomics Protocols; Peacock, C., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1201, pp. 339–361. ISBN 978-1-4939-1437-1. [Google Scholar]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [Green Version]
- Dumetz, F.; Imamura, H.; Sanders, M.; Seblova, V.; Myskova, J.; Pescher, P.; Vanaerschot, M.; Meehan, C.J.; Cuypers, B.; De Muylder, G.; et al. Modulation of Aneuploidy in Leishmania donovani during Adaptation to Different In Vitro and In Vivo Environments and Its Impact on Gene Expression. mBio 2017, 8, e00599-17. [Google Scholar] [CrossRef] [Green Version]
- Lypaczewski, P.; Thakur, L.; Jain, A.; Kumari, S.; Paulini, K.; Matlashewski, G.; Jain, M. An Intraspecies Leishmania Donovani Hybrid from the Indian Subcontinent Is Associated with an Atypical Phenotype of Cutaneous Disease. iScience 2022, 25, 103802. [Google Scholar] [CrossRef]
- Leprohon, P.; Legare, D.; Raymond, F.; Madore, E.; Hardiman, G.; Corbeil, J.; Ouellette, M. Gene Expression Modulation Is Associated with Gene Amplification, Supernumerary Chromosomes and Chromosome Loss in Antimony-Resistant Leishmania Infantum. Nucleic Acids Res. 2009, 37, 1387–1399. [Google Scholar] [CrossRef] [Green Version]
- Engwerda, C.R.; Kaye, P.M. Organ-Specific Immune Responses Associated with Infectious Disease. Immunol. Today 2000, 21, 73–78. [Google Scholar] [CrossRef]
- Debrabant, A.; Joshi, M.B.; Pimenta, P.F.P.; Dwyer, D.M. Generation of Leishmania Donovani Axenic Amastigotes: Their Growth and Biological Characteristics. Int. J. Parasitol. 2004, 34, 205–217. [Google Scholar] [CrossRef]
- Bates, P.A. Leishmania Sand Fly Interaction: Progress and Challenges. Curr. Opin. Microbiol. 2008, 11, 340–344. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence |
|---|---|
| LdSAcP1 | 5′ ATCGAAGACCTTTGTCTTCGCCGTTACCATCTTCGGTTTTAGAGCTAGAAATAGCAAG |
| LdSAcP2 | 5′ ATCGAAGACCCAAACCAAATCGGCCGTTCCTATCACCATGACGAGCTTACTC |
| LdSAcPBleF | 5′ TCTGCGTCCCACCGGAATCCCTCAAGATCTTCATCGGATCGGGTA |
| LdSAcPBleR | 5′ GCGGTGTCCTGGAGCAGCTCCACGAGTCGGTCAGTCCTGCTCCT |
| LdSAcPF2 | 5’ CCGCCCACTCAACGATTAT |
| LdSAcPR | 5′ CTGTACTGAGCCTGCGTCAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulini, K.; Lypaczewski, P.; Zhang, W.-W.; Perera, D.J.; Ndao, M.; Matlashewski, G. Investigating the Leishmania donovani sacp Gene and Its Role in Macrophage Infection and Survival in Mice. Trop. Med. Infect. Dis. 2022, 7, 384. https://doi.org/10.3390/tropicalmed7110384
Paulini K, Lypaczewski P, Zhang W-W, Perera DJ, Ndao M, Matlashewski G. Investigating the Leishmania donovani sacp Gene and Its Role in Macrophage Infection and Survival in Mice. Tropical Medicine and Infectious Disease. 2022; 7(11):384. https://doi.org/10.3390/tropicalmed7110384
Chicago/Turabian StylePaulini, Kayla, Patrick Lypaczewski, Wen-Wei Zhang, Dilhan J. Perera, Momar Ndao, and Greg Matlashewski. 2022. "Investigating the Leishmania donovani sacp Gene and Its Role in Macrophage Infection and Survival in Mice" Tropical Medicine and Infectious Disease 7, no. 11: 384. https://doi.org/10.3390/tropicalmed7110384