1. Introduction
Tribosystems in nature, such as the oral cavity, the corneal epithelium and the articular cartilage provide exceptional life-long lubrication [
1]. Biological tribosystems exhibit a biphasic composition, comprised of a macromolecular network consisting of mucins [
2,
3], polysaccharides [
4], phospholipids [
5], or glycoproteins [
6], and a water-based lubricant [
1]. Due to their compositional resemblance to biotribosystems, along with their hydrophilicity, biocompatibility and tunability of microstructure and chemical composition [
7,
8,
9], hydrogels have gained popularity since their inception in 1960, both to study mechanisms underlying biolubrication, and as biomaterials, e.g., for cartilage replacement [
10].
Figure 1 shows a schematic representation of a hydrogel’s structure. Hydrogels are composed of chemically or physically crosslinked polymeric networks imbibed by a good solvent [
11], in most cases an aqueous fluid. The blob-like structure of hydrogels is often described via the scaling laws proposed by de Gennes for polymer solutions in the semi-dilute regime. The distance between two crosslinks (labelled as mesh size,
) depends on the polymer concentration and the solvent quality [
12,
13], among others. This characteristic length determines key properties of hydrogels like permeability, elastic modulus and viscoelastic relaxation (
Table 1).
Numerous studies have experimentally explored the load and speed dependence of the frictional response of hydrogels. Studies pioneered by Gong et al. showed that hydrogel friction does not follow Amonton’s law, i.e.,
, where
is the friction force,
is the normal load, and
the friction coefficient [
16]. Subsequent studies showed that friction can either increase with load [
17], be load-independent [
16], or even decrease with increase in load [
18,
19]. A prominent dependence of friction on the sliding speed was reported concurrently and associated with viscous dissipation. This was initially attributed to the strong hydration of the gel, and thereby, to the possibility of sustaining hydrodynamic lubrication even at low sliding velocity and relatively high contact pressure. A dependence of friction on sliding velocity has been reported in many subsequent studies and over a wide range of conditions. Although these studies have also shown that hydrogel friction also varies with the chemical composition of the polymer [
20,
21,
22], monomer and crosslinking concentrations [
23,
24], and surface roughness [
25,
26], a detailed description of these results is out of the scope of this review.
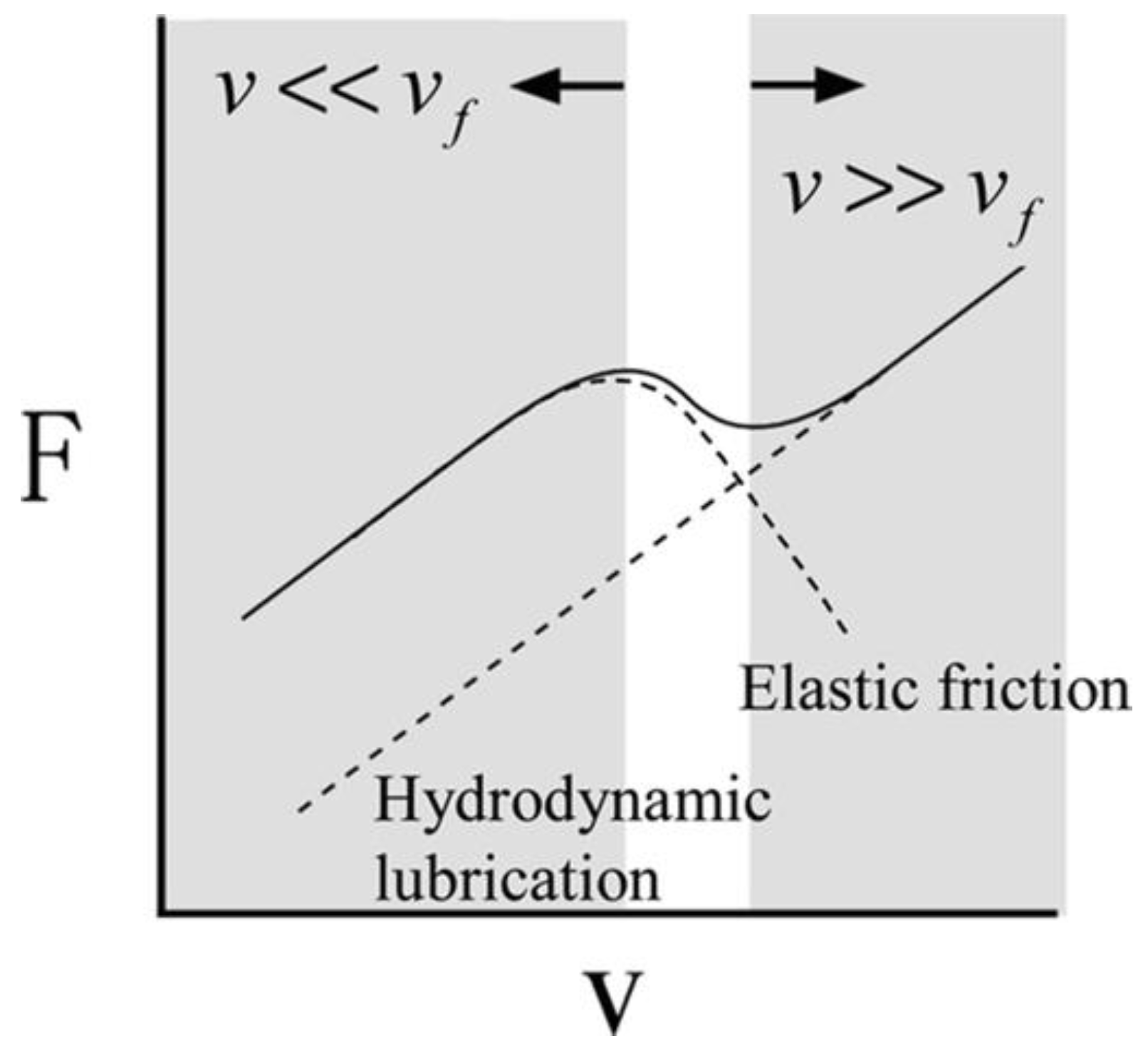
A plethora of mechanisms have been proposed to explain the low friction coefficient provided by hydrogels. Commonalities among tribological studies include the relevance of distinct lubrication mechanisms at low and high sliding velocities. At high sliding velocities, friction results from the viscous dissipation upon shear of a fluid. Both hydrodynamic and non-hydrodynamic dissipative mechanisms arising from viscoelastic deformation have been proposed. At low sliding velocities, the frictional characteristics strongly depend on the adhesive vs. repulsive nature of the contact. It was Gong who first made the distinction between adhesive and non-adhesive (repulsive) contacts with hydrogels [
27]. Additionally, the contact type (e.g., migrating vs. stationary contact) also plays a role in dictating frictional mechanism. In migrating contacts (e.g., a hard pin sliding on a hydrogel flat surface), a re-hydration of the interface and polymer relaxation are possible during the stress-free periods, while stationary contacts (e.g., a hydrogel microsphere sliding along a solid surface or a ring-on-disc tribometer) do not enable re-hydration of the near-surface region of the hydrogel (
Figure 2). At small enough stroke lengths, reciprocating and unidirectional sliding have led to similar frictional characteristics of investigated hydrogels [
28]. However, if the hydrogel surfaces have a texture or the surface structures are oriented/aligned in a particular direction, the sliding direction could affect the frictional response.
The goal of this review was not to cover all of the literature on hydrogel lubrication but to discuss specific models that have attempted to quantify hydrogel friction. More specifically, this review is mainly focused on adhesive contacts between a solid surface and a hydrogel, although some findings obtained on repulsive contacts are presented to explain available models for viscous dissipation. The paper is organized in three main sections. First, the main models for hydrogel friction are described. One subset of models takes a polymer physics approach based on the scaling laws proposed by de Gennes [
13], and emphasizes the role of the mesh size both in boundary and in hydrodynamic lubrication. In contrast to this, the second type of models emphasizes the role of a thin fluid film effectively separating the countersurfaces at low velocities and/or full fluid film lubrication at fast sliding velocities. This paper discusses these apparently contradictory models for single network hydrogels and their conditions of validity. This is followed by a detailed description of our own model for hydrogel friction and its application to two different hydrogels: polyacrylamide and agarose hydrogels, which lead to adhesive and repulsive contacts with a solid surface, respectively. We conclude with closing remarks and opportunities for future research to the best of the authors’ knowledge.
3. Elastic Contribution to Frictional Dissipation
Gong’s studies of hydrogel lubrication significantly contributed to advancing the knowledge of mechanisms underlying hydrogel friction [
20,
21,
22,
27,
50]. The main idea is that friction results from two major contributions, the energy dissipated due to the rupture of adhesive bonds across the interface and the viscous dissipation originated by the shear of the solvent film separating the two surfaces at sufficiently high sliding velocities [
20]. Gong’s model for adhesive contacts [
27] assumes an intermittent and unconcerted adsorption and desorption of polymer chains to the solid surface. As the countersurface moves, the pinned polymer strands stretch, thereby storing an elastic force,
:
where
is the sliding velocity,
the contact time, and
the thermal energy per polymer chain. When detachment occurs, the stored elastic energy is dissipated. The elastic contribution to hydrogel friction depends on the relation between contact time (
) and the relaxation dynamics, which are characterized by the lifetime of the adsorbed chain (
) and the polymer relaxation time
(
). At the interface,
represents the time for re-attachment of the polymer after detachment. The model predicts an increase in friction with sliding velocity due to the increasing number of adhesive bonds—often labelled as velocity-strengthening friction. Because the lifetime decreases with an increase in velocity, the adhesive frictional force exhibits a decreasing trend at sufficiently high velocities—or velocity-weakening regime—and a peak or plateau in between (
Figure 4). Gong observed that weakly adhesive contacts showed a peak in friction at a velocity
(
in
Figure 4) [
51]. At sufficiently high sliding velocities, hydrodynamic lubrication was assumed to dominate over the contribution of the elastic friction.
Improvements of the adsorption–desorption model have been proposed, e.g., the so-called Population Balance equation (PBE) [
52]. Instead of the Hookean spring model to calculate
, the PBE model includes a finitely extensible non-linear elastic model for the stretching of the polymer. In addition, a viscous retardation stress accounts for the relative motion of the polymer chains surrounding the stretched polymer chain. Furthermore, it considers an average bond age
that differs from its lifetime
. Gupta et al. applied the PBE model to the friction of gelatin hydrogels [
53]. The model was able to predict the increasing friction with gelatin concentration (and decrease in mesh size) originating from the enhanced adhesive bonds at the interface. Furthermore, the model was used to derive scaling laws of frictional parameters. An interesting result is that the shear modulus was observed to depend on the sliding velocity. This is an inherent property of a viscoelastic material, which behaves more fluid-like at low shear rates and more solid-like at higher shear rates. The work also showed that the Hookean approximation is valid for hydrogels, mainly because of the weak interactions at the interface. Nevertheless, if the binding interactions were to be strong (e.g., via electrostatic interactions), the polymer chains could be extended beyond the linear regime.
An adhesive model was also proposed by Baumberger for gelatin–glass tribopairs [
54,
55]. As in Gong’s model, the hydrogel surface consists of polymer blobs with a characteristic size close to
, and the blobs adhere to the glass surface due to van der Waals interactions, electrostatic attractions or hydrogen bonds. Baumberger’s studies demonstrated the scaling of friction with the surface density of adhesive blobs, in marked contrast to the mechanism of hydration lubrication for repulsive contacts. This was further supported by observations of intermittent sliding or stick-slip due to the periodic attachment (detachment) of the network to (from) the countersurface, which was denoted as “self-healing slip pulses” [
54]. Here, frictional slip occurs by propagating pulses that can self-heal (re-pin), by analogy to intermittent fault dynamics during an earthquake [
56]. At velocities below a critical value, re-pinning occurs at the pulse’s trailing edge and at once throughout the contact. The velocity of the slip or fracture front (
) was measured and found to depend on the mesh size and the collective diffusion coefficient of the polymer network (
) as
[
55]. Once the velocity exceeds a critical value, adhesive bond formation is insignificant, and the sliding becomes smooth. Here, the authors proposed that friction originates from the shear of a polymer solution film having a thickness comparable to that of the mesh size of the hydrogels and exhibiting shear thinning behavior; this is discussed later in detail. It is noteworthy that this work also introduced the concept of “aging”, namely that the binding strength at the glass-hydrogel interface depends on the contact time.
3.1. The Concept of Critical Velocity
A common concept in Baumberger and Gong’s works is the existence of a critical velocity at which the lubrication mechanism transitions to viscous dissipation. This critical velocity has been related to the dimensionless Weissenberg number (
), which is classically used to describe viscoelastic flow. The Weissenberg number compares elastic and viscous forces via the ratio between a relaxation time and an experimental time (
). If the relaxation time of the polymer (
) is equal to the interaction time with the counter-surface (
), elastic and viscous forces are of similar relevance. This happens at a critical velocity
, where
[
27,
50]. If
, elastic forces are more relevant than viscous forces (
, low
-numbers), and
vice versa if
.
Table 2 shows the critical velocity
that we calculated for the experiments in previous works. The magnitude of the transition velocity (
) was extracted from the reported data, considering that viscous dissipation happens at the highest sliding velocities. As
Table 2 demonstrates,
is various orders of magnitude smaller than the calculated critical velocity,
. It is intuitive that polymer chains are free to fluctuate at hydrogel-hydrogel interfaces. However, under the confinement provided by a solid surface in an adhesive contact, the polymer relaxation in the near-surface region should be restricted to some extent when water is drained, as reported for polymer brushes [
38]. In addition, the critical velocity was estimated with the mesh size of the bulk hydrogel
. However, recent studies have demonstrated that the near-surface region of hydrogels may have structural characteristics that differ from the bulk as a result of the modified polymerization reaction close to the interface. All this is expected to contribute to the deviation between
and
shown in
Table 2.
3.2. Relevance of Microstructural Gradients
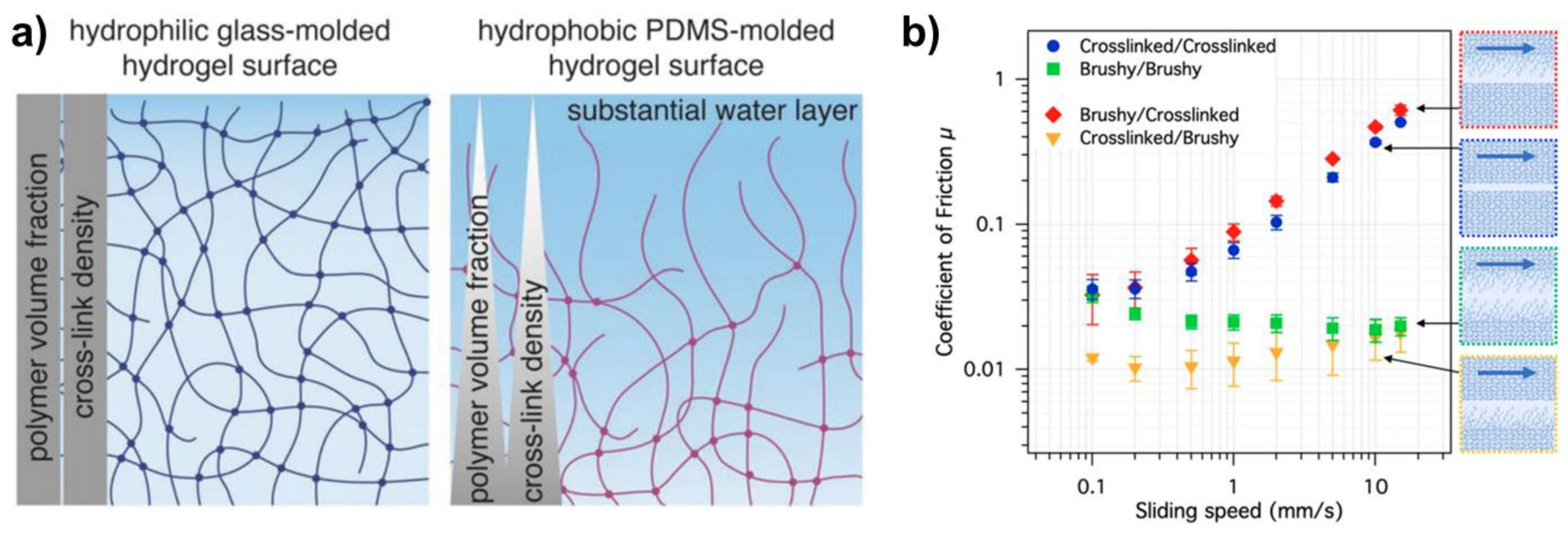
A synthesis-sensitive graded microstructure has been reported for polyacrylamide hydrogels [
27,
44,
59]. The hydrogel’s surface, when molded against a hydrophobic surface, results in a soft, highly hydrated and loosely crosslinked (“brush-like”) near-surface region (
Figure 5a). A brush is characterized by its height (
) [
60]. The relaxation of dangling chains differs from the relaxation of the crosslinked polymer network, which diffuses in a cooperative motion. This brush-like region can have a thickness as large as ~4 μm [
44]. For example, Shoaib et al. found that PAAm hydrogels with 4, 6 and 9 wt % monomer and 0.1, 0.3 and 0.45 wt % crosslinker, when molded against hydrophobic glass slides treated with dichloro dimethylsilane, have a loosely crosslinked surface layer with an approximate thickness of ~640, 330 and 250 nm, respectively [
32]. We discuss results for these hydrogels later.
The influence of the brushy surface region on the friction coefficient is remarkable (
Figure 5b). Its presence reduces friction significantly. It can also be expected that the existence of this surface layer affects
. In fact, the fits of the viscous-adhesive model to the measured friction force between PAAm hydrogels and a silica sphere [
23] lead to relaxation times for attachment/detachment of the polymer to/from the countersurface that are much longer than expected for the cooperative diffusion of blobs of size
. This might be caused by the contribution of the characteristic length of the near-surface region,
, instead of the mesh size of the bulk gel. Halperin [
61] estimated the longest relaxation time required for a tethered polymer chain to stretch or collapse a distance equal to its thickness by the reptation time,
.
Table 3 shows that the relaxation times estimated for
and
for three different polyacrylamide hydrogels differ by about 4–5 orders of magnitude, with
. Accordingly, very different relaxation dynamics can be expected for the polymer at the interface. Please note that this estimation neglects that the applied pressure can yield the drainage of water and an increase in polymer concentration, as well as that the polymer chains can be crosslinked on both ends, all of which can further slow down the polymer dynamics.
4. Poroelastically Induced Drainage of Water and Induced Frictional Dissipation
Pressure-driven fluid flow in the hydrogel occurs only when the applied pressure exceeds the osmotic pressure of the hydrogel,
[
62], and can also be the origin of frictional dissipation. The drainage of water upon indentation of the hydrogel with rigid probes has been described within the framework of poroelastic theories, which couples the elasticity of the gel network with the pressure-induced flow of water. The footprint of a poroelastically induced dissipation is a decrease in friction with an increase in sliding velocity, as the time for fluid drainage and the dissipation are reduced. This is often described in terms of a Peclet number (Pe), which gives the relation between diffusive and advective time,
, where
is the poroelastic relaxation time,
the sliding velocity, and
the contact radius. The value of
can be roughly estimated with the hydrogel diffusivity (
Table 1), and depends on the mesh size, the elastic modulus of the hydrogel, and the applied pressure. Large Peclet numbers (large
and fast sliding velocities) are thus associated with lower friction coefficients.
Several analytical models have been derived for the poroelastically induced frictional dissipation [
58,
63,
64]. Interestingly, Delavoipière et al. [
62] identified two different regimes by visualizing the contact area during sliding through reflection interference contrast microscopy. In the low-velocity regime (i.e., when Pe < 1), the contact line remains circular (ζ ~ 1) with a constant radius close to the radius achieved under static indentation loading at the same normal force (
a0). Here, friction increases with sliding velocity (
Figure 6). In the high-velocity regime (Pe > 1), the contact radius decreases progressively with increasing sliding velocity while the asymmetry parameter ζ first decreases and then increases. This change in the shape of the contact area was demonstrated not to result from a hydrodynamic lift but from a pressure imbalance between leading and trailing edges arising from the poroelastically driven fluid flow, resulting in a lifting force. Most of the frictional dissipation was accounted for by a fracture mechanics approach, where the leading and trailing edges of the contact were viewed as closing and opening cracks, respectively, and dissipation arose from the poroelastic flow. Instead of considering fracture mechanics, the main idea in Reale and Dunn’s for their poroelastically driven lubrication model is that a decrease in velocity promotes dehydration of the contact, which yields an increase in adhesion, and thereby, in friction. Accordingly, they modeled the friction coefficient as a function of the Peclet number and the interfacial energy [
58].
The findings by Delavoipière allow the identification of the experimental conditions at which poroelastically induced friction is of relevance.
Table 4 shows the contact times (
as well as the poroelastic relaxations times,
, calculated with the tribological parameters provided in several reported experiments.
Table 4 shows multiple studies in which poroelastic drainage is of relevance. This is, of course, largely dependent upon the experimental conditions such as the sliding velocities and the contact pressures. In works by Reale and Dunn [
58] and Delavoipière [
64], poroelastic drainage is relevant under many experimental conditions, excluding the highest speeds probed (highest Pe numbers). In Ref. [
23], the poroelastic drainage in PAAm-4%, PAAm-6% and PAAm-9% hydrogels is relevant, which mainly results from the small contact size in AFM experiments.
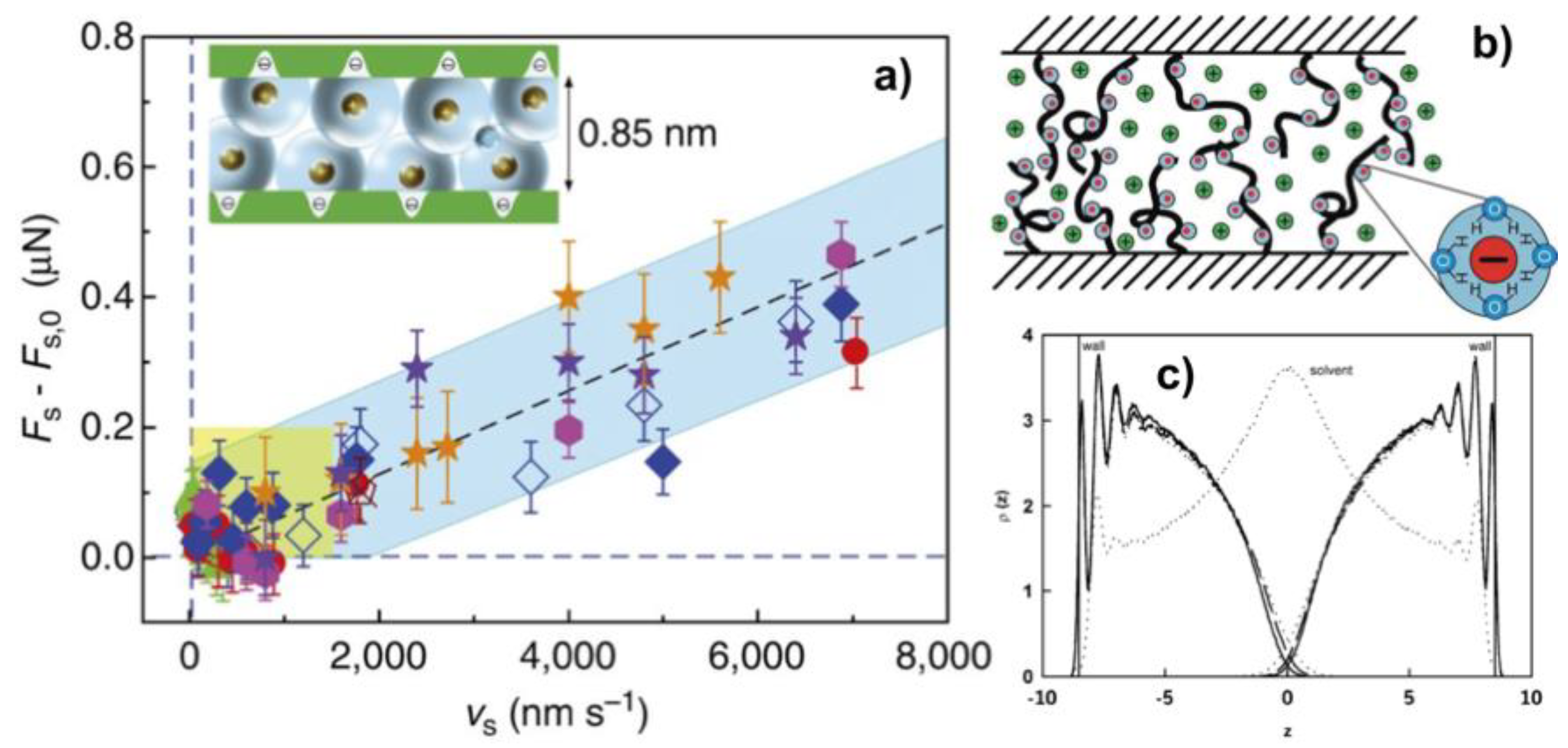
We also emphasize that the presence of microstructural gradients should affect the poroelastic relaxation. Depending on the stress distribution, the pressure-induced drainage of water can happen through the near-surface region with lesser resistance to flow in case of lower crosslinking degree, and/or through the bulk network. For instance, PAAm-6% hydrogels have a surface characteristic length ( of 350 nm and a bulk mesh size ( of ~8.2 nm, and elastic moduli of 455 and 8700 Pa, respectively. Upon indentation with a colloid (R = 10 μm, L = 10 nN), the corresponding poroelastic times are = 1.23 s and = 36 s for the surface and the bulk, respectively, hence resulting in an order of magnitude difference in poroelastic relaxation. Although a precise quantification is difficult because the stress field is unknown, it is important to consider that the poroelastic relaxation time may be significantly altered by the presence of microstructural gradients.
5. Viscous Dissipation
Hydrodynamic lubrication occurs when the sliding velocity is sufficiently high that there is a hydrodynamic lift, which yields a sustained fluid film separating the two surfaces in relative motion. The motion of a solid in a viscous fluid generates a shear stress in the fluid and brings it in motion. The viscous dissipation arises from the irreversible transformation of the work done by the shear in the fluid into thermal energy. With an increase in sliding velocity, the dissipated energy increases while this fluid film becomes thicker. Note that this deviates from the mechanism of hydration lubrication proposed by Klein and discussed earlier, which relies on the steric and osmotic repulsion provided by polymer brushes for the formation of a thin fluid film at the interface.
According to Newton’s law of viscosity and taking into account de Gennes’ scaling theory [
66], Gong proposed the following expression for the friction force in the hydrodynamic regime:
where
is the polymer concentration,
the applied pressure and
the contact area between parallel plates with a relative sliding velocity
. Experimental results showed, however, a sublinear relation between
and
,i.e.
with
~ 0.21–0.55 [
20,
51]. Hence, this expression was modified to account for the separation between the two surfaces and the Newtonian flow of the solvent within the gel, introducing the permeability
of the hydrogel and its relation to the mesh size [
20]. However, despite these improvements, the model underestimated the measured friction force by one order of magnitude. To justify this, the existence of bound water in the proximity of the polymer moieties and/or an increase in viscosity of the confined water was speculated [
20]. This assumption is, however, doubtful, because the film thickness was estimated to be larger than 10 nm and an increase in the viscosity of water has not been demonstrated yet for such thick films. Furthermore, previous works have suggested that confined water retains fluidity under nanoconfinement [
38].
Several experimental works have confirmed the sublinear relation
. For example, Kagata et al. [
51] showed that the exponent
increases from ~0 to ~0.55 with an increase in pressure, while the velocity dependence is less prominent (i.e., lower values of
) at slow sliding velocities. Without further proof, a non-Newtonian behavior of water and non-hydrodynamic dissipative mechanism were proposed to be responsible for the experimental results. A sublinear relation
has also been reported for adhesive contacts above a transition or critical velocity [
23,
51,
54,
67]. Understanding the discrepancy among the reported exponents (
from ~0.02 to 0.7) and its load dependence is still the subject of research to date [
20,
23,
28,
35,
51,
67].
One of the shortcomings of Equation (2) is that it neglects the hydrodynamic variation of the film thickness with velocity arising from the deformation of the hydrogel. The soft hydrogel is deformed by the lubrication pressure generated in the thin film, leading to a net lift force that maintains the film [
29,
64,
68]. For the case of an isoviscous–elastic contact, Hamrock and Dowson [
69] proposed a viscous shearing force as
, yielding
, which is qualitatively in better agreement with experimental results. Ureña et al. [
67] showed, however, that the elastohydrodynamic theory underestimates the friction coefficient between two polyacrylamide hydrogel surfaces, and hence, a quantitative agreement is still lacking. The observed discrepancy could arise from neglecting the permeable nature of the hydrogels or from the geometrical differences between a ball-on-flat and a flat-on-flat contact, but a precise analysis has not been performed yet.
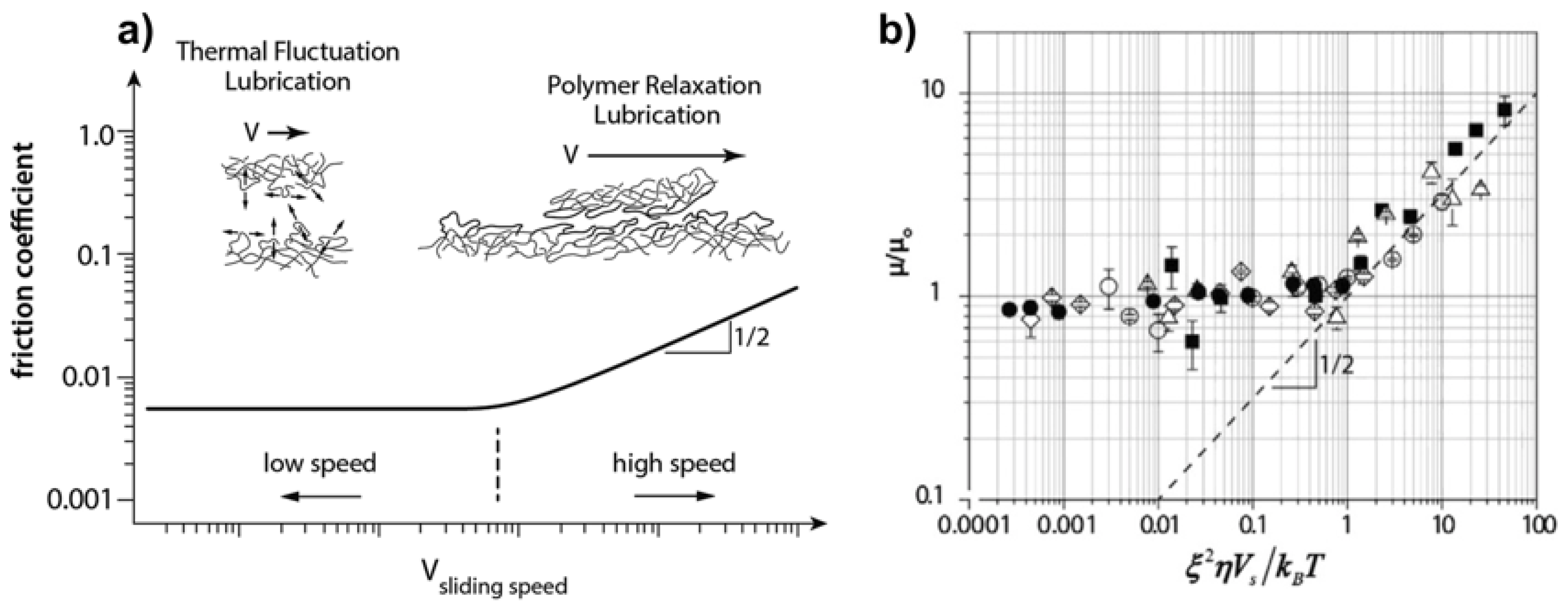
A transition from a low, speed-independent friction coefficient (at slow sliding speeds) to a speed-dependent friction coefficient
was observed for Gemini hydrogel (repulsive) interfaces by Sawyer’s group (
Figure 7b) [
57,
70]. Although an exponent of 0.5 could be predicted by an elastohydrodynamic approach, the predicted friction coefficient was more than one order of magnitude lower than the measured values. The authors proposed “thermal-fluctuation lubrication” as the underlying mechanism at slow sliding velocities. That is, random thermal chain fluctuations—of length
—at the interface relax the shear stress generated during sliding and provide a blurred interface over which the barrier to sliding is effectively reduced compared to a hard interface. This is reminiscent of the polymer fluctuations shown in MD simulations of polymer brushes under shear [
45]. The transition to higher friction coefficients at high sliding velocities was not attributed to full-fluid film (hydrodynamic) lubrication. Instead, it was proposed that the polymer relaxation was still involved in the lubrication of the repulsive contact (
Figure 7a). However, the exact mechanism was not elucidated [
31,
57]. More recently, Simič et al. [
28] proposed that the weak dependence of friction on velocity at slow sliding velocities in repulsive contacts reflects brush-like lubrication, i.e., hydration lubrication, and hence, it is dominated by the shear of the solvent.
A recent comprehensive study of the friction of polyacrylic acid (PAA), PAAm and agarose hydrogel spheres sliding on smooth solid surfaces yielding repulsive contacts has emphasized the relevance of hydrodynamics in hydrogel friction [
29]. Based on the relation between the measured friction coefficient and the hydrogel mesh size, the friction force at low velocities is consistent with the dissipation by a hydrodynamic flow through the porous hydrogel network:
This differs from the brush-like hydration lubrication proposed by Spencer [
28] or the thermal fluctuation lubrication suggested by Sawyer [
57]. It is noteworthy, though, that the mesh size was not experimentally determined, but approximate values were assumed based on other works, and hence, perhaps only a qualitative comparison to the experimental results is granted here. At high velocities, a mesoscopic lubricating liquid film was presumed to form between the hydrogel and the solid surface. Agreement with elastohydrodynamic theory was only observed for the softest hydrogels, and the origin of the discrepancy for the stiffer gels was not elucidated. Interestingly, the frictional force decreased by an order of magnitude between these two regimes and displayed slow relaxation over several minutes (
Figure 8). This was interpreted as an interfacial shear thinning of the polymer due to its alignment in the shear direction within a confined volume, which reduced friction before the onset of hydrodynamic lubrication. The observed time dependence of the friction force in this intermediate regime was justified by the effect of confinement on prolonging the timescale required for the polymer to explore the configurational space. Kim and Dunn modeled the shear stress in this regime using a thixotropic flow model to represent the time-dependent structural changes of the interface [
71]; however, instead, they attributed the time-dependent frictional response to transient interfacial rehydration. Rehydration could not explain Cuccia et al. results [
29], though.
It is interesting that the time-dependent intermediate regime was only prominent in static contacts in Ref. [
27]. Perhaps this is because the polymers near the hydrogel surface in a migrating contact only undergo shear loading transiently, which might hinder the shear-induced alignment and the confinement of entanglements compared to the situation in static contacts.
The idea of a non-hydrodynamic dissipative mechanism at high sliding velocities was elaborated by Baumberger et al. [
55] for gelatin hydrogels and a glass countersurface yielding an adhesive interface. It was proposed that friction stems from the shear of a hydrogel film with a thickness of roughly the mesh size. The effective viscosity
of this superficial region was defined as:
where
is the shear rate and
the shear stress. Assuming that the behavior of the polymer chains follows that of the Rouse model [
72], the relaxation was determined as
, where
is the monomer size. The Rouse chain model assumes a dilute polymer solution below its point of entanglement, and hence, it deviates from the classical picture of a crosslinked network. Interestingly, it qualitatively agrees with the picture of a brush-like superficial layer for hydrogels with a graded microstructure (
Figure 5a). The Weissenberg number (
) was determined as the ratio between the stress relaxation time (
) and an experimental time ~
.
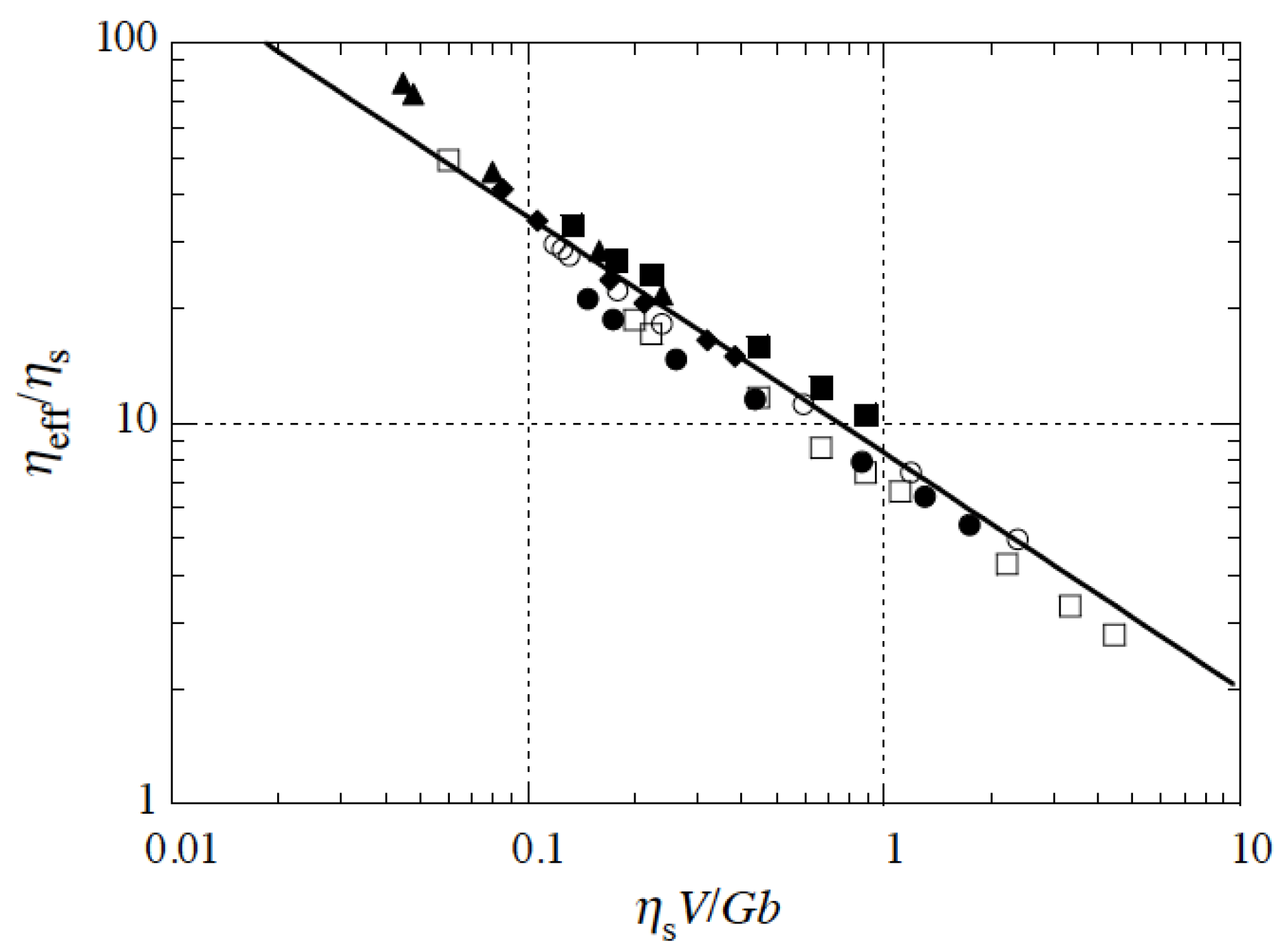
Figure 9 shows how the experimental data corresponding to hydrogels with varying mesh size collapse onto a master curve
. This indicates that the mesh size influences the viscous dissipation and that
, thereby supporting the shear-thinning behavior of the superficial region of the gelatin hydrogels when sheared against a glass surface.
6. The Viscous-Adhesive Model for Hydrogel Friction
The viscous-adhesive model was introduced in Ref. [
23] by the authors of this review to describe the frictional characteristics of the adhesive interface between polyacrylamide hydrogels and a silica microsphere. As in Schallamach’s model for rubber friction [
73] and in Gong’s adsorption-desorption model, energy dissipation arises from the shear-induced rupture of the transient adhesive bonds (i.e., junctions) formed at the colloid-hydrogel interface. Our approach is conceptually similar to Schallamach’s; however, the effect of the shear-assisted decrease on the energy barrier for bond rupture is neglected due to the small magnitude of the friction force. This enables us to derive a simple analytical expression for the adhesive (elastic, according to Gong’s nomenclature) contribution to friction (
).
is thus given by the strain of the junction (
) multiplied by its shear modulus (
) and by the area of the adhesive junctions (
):
with
being the time elapsed since the zero-state stress,
the sliding velocity,
the length of the polymer junction,
the bond rupture time, and
the yield length (so
is the yield strain). The yield strain considers that rupture can also happen at the yield length, a concept introduced by Drummond et al. [
74] for surfactant monolayers. The area in adhesive or pinned state (
) is estimated from
, with
being the migrating contact area (
),
the characteristic time of bond formation and
the mean life time of the adhesive junction,
=
, which accounts for the probability that a junction is in adhesive state. Equation (5) predicts that
can increase and decrease with
and a peak or plateau can be achieved in the intermediate regime. The adhesive contribution to friction depends on characteristics of the polymer network at the confined interface (
,
,
,
and
). The derivation of Equation (5) is described in detail in the SI of Ref. [
23].
The sliding contact area
takes into account the poroelastically induced deformation of the hydrogel in colloidal probe AFM experiments, and hence, it can be smaller than the static contact area
. In the limit of small deformations, a sliding contact radius
is estimated from:
being the poroelastic relaxation time of the hydrogel with an effective diffusivity
. We distinguish between
and
for migrating (subindex “V”) and static contacts (subindex “0”), respectively; similarly,
and
are the indentation depths of the migrating and static contacts.
is assumed to be the same for both and
is the contact time (
and
of the static and migrating contact, respectively). The indentation depths,
and
, are described via a Kelvin–Voigt model, appropriate for polyacrylamide hydrogels [
75]. Linearization of this expression leads to:
Rearranging the above expression, the velocity-dependent contact radius
and contact area
are given as:
Several works have attempted to visualize the migrating contact area to obtain a more precise estimation [
29,
64,
76]; see, e.g.,
Figure 6. The work by Delavoipière visualized a decrease in contact area (
) with velocity at Peclet numbers larger than 1.
On the other hand, the resistance to the motion of a solid (the silica microsphere) in a fluid leads to viscous dissipation, which constitutes the viscous contribution to friction. Here, we use a general expression for the viscous drag,
where
is the effective viscosity of the fluid and
is a geometric factor. For a plane–plane geometry,
, where
is the thickness of the sheared film and can include the hydrogel;
might also depend on the sliding velocity, but this is neglected here. For a sphere–plane geometry like the AFM geometry in
Figure 2d,
[
77]. The WLF equation, which has worked well to describe the shear-thinning behavior (
< 1) of nanoconfined polymers [
77], is used to model the effective viscosity,
. In the case of a Newtonian fluid (
),
should be equal to the viscosity of the film
. If the polymer influences the viscous dissipation, then the viscosity
will deviate from the solvent viscosity,
.
The kinetic friction is given by
where
is a velocity-independent term, which was observed in experiments [
23]. Our more recent results on agarose hydrogels suggest that it might be of viscous origin, as argued later.
The minimization of
yields an expression for the transition velocity
at which
achieves a minimum in friction. The simplest form of
is obtained for parallel plates:
Based on this model, the transition velocity (
) depends on the elasticity of the transient junctions,
, the yield length
, the time for the formation of adhesive bonds at the confined interface
, and the viscosity parameters
and
of the rheological model. Hence, this expression demonstrates that
arises from the competition between adhesive and viscous contributions to friction, like
. However, there are obvious differences between the expressions for
and
. First,
. accounts for non-Newtonian behavior, if present. Second,
can deviate from
, e.g., if the confinement provided by the solid surface slows down polymer dynamics. Third, it considers that rupture can happen above a yield length
, and hence, it is not only dictated by the fluctuation dynamics of the polymer. We find that the observed transition velocity in our experiments [
23] is of the same order of magnitude as the calculated velocity
(see
Figure 10) and much smaller than
(see
Table 2). The discrepancy is mainly due to the prolonged relaxation time, i.e.,
.
The viscous-adhesive model was used to describe the velocity and load-dependent friction force between a silica colloid and polyacrylamide hydrogels measured by AFM in Ref. [
23]. Polyacrylamide can form hydrogen bonds with silica [
78], and hence, the contact is adhesive. Here, we compare these results to the frictional response of agarose hydrogels, which exhibit a much weaker adhesion to the silica colloid. Agarose hydrogels were prepared by dissolving 1 wt % of agarose powder in DI water at a temperature of 80 °C and continuous stirring at 375 rpm. After dissolution, 2 mL of the solution was pipetted into circular molds and left to gelate at room temperature for 30 min. Then, DI water was added to the petri dishes, and gel samples were stored inside the fridge overnight. All gels were tested on the next day. The gel samples were rinsed with DI water to remove the uncrosslinked monomer. Polyacrylamide hydrogels were prepared with three different concentrations of the monomer, as reported in detail in our previous works [
23,
24,
32]. All the measurements shown in this section were conducted with DI water as the solvent.
For the selected compositions of PAAm hydrogels,
Table 5 shows the elastic moduli and the adhesion energy as measured by colloidal probe AFM, as well as the shear storage modulus as measured with a rheometer. The investigated agarose hydrogels exhibit an elastic modulus of 2.45 ± 0.99 kPa and zero adhesion energy under the same experimental conditions.
Unknown parameters in Equations (5) and (6) are the thickness of the junction (
) and the thickness of the shear fluid film (
), respectively. In Ref. [
23], we assumed them to be equal to the indentation depth. Here, inspired by Baumberger’s and Cuccia’s works [
29,
55], we assume that both
and
are close to the correlation length of the hydrogel in the near-surface region. However, this deviates from the mesh size of the bulk hydrogel (
) due to the graded microstructure of our hydrogels. Hence, we assume here that both
and
are equal to the height of the brush-like surface region (~
); see
Table 3.
Fits of the viscous-adhesive model to the experimental results provide the magnitude of the fitting parameters. It is evident in
Figure 11 that the friction force is higher for PAAm-9% hydrogels. According to our model, this is mainly because the characteristic time for the formation of adhesive bonds (
) is notably smaller for PAAm-9% hydrogels compared to the other hydrogels, while the time for bond rupture (
) is only marginally smaller than that of PAAm-6% hydrogels (
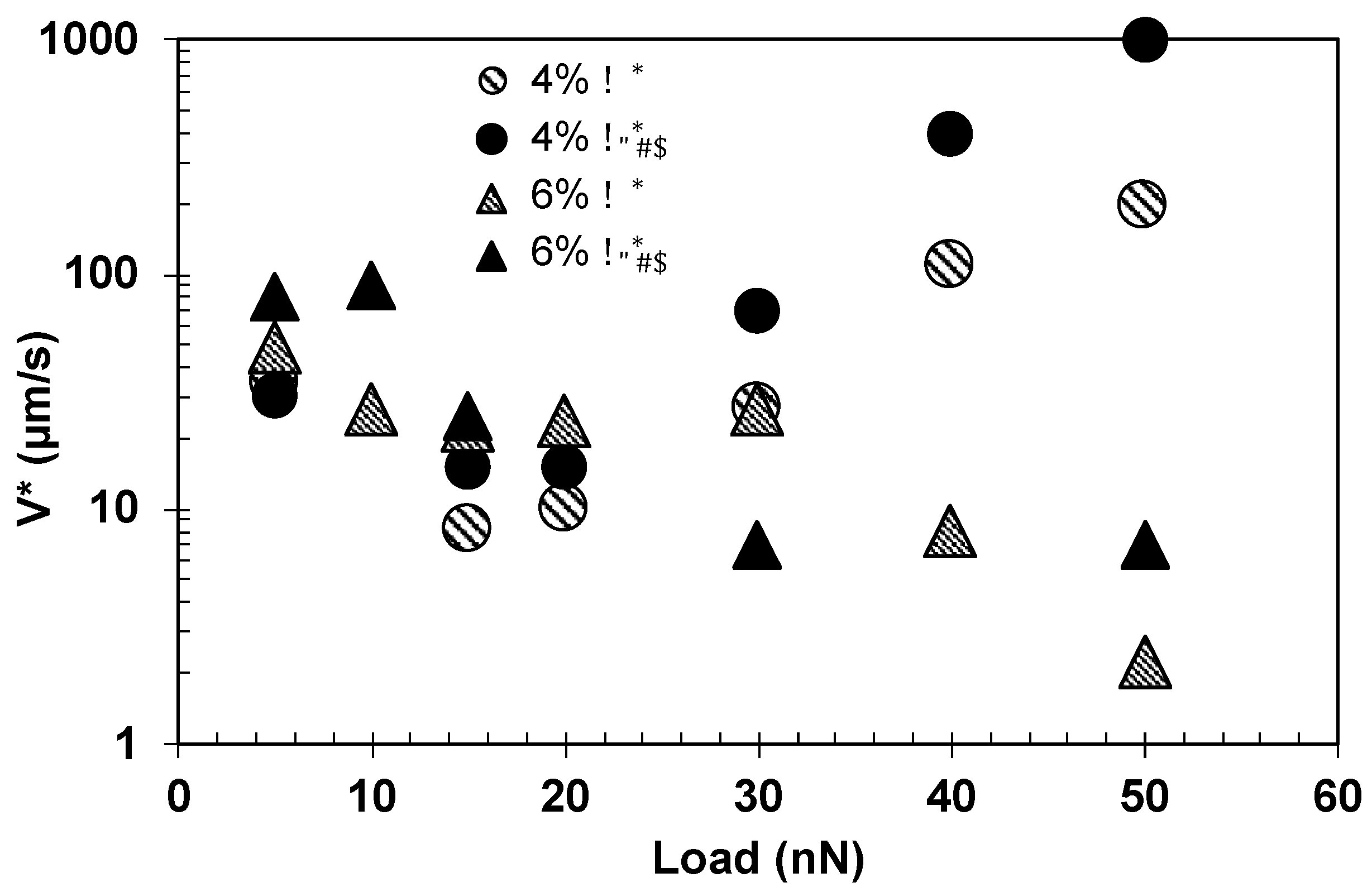
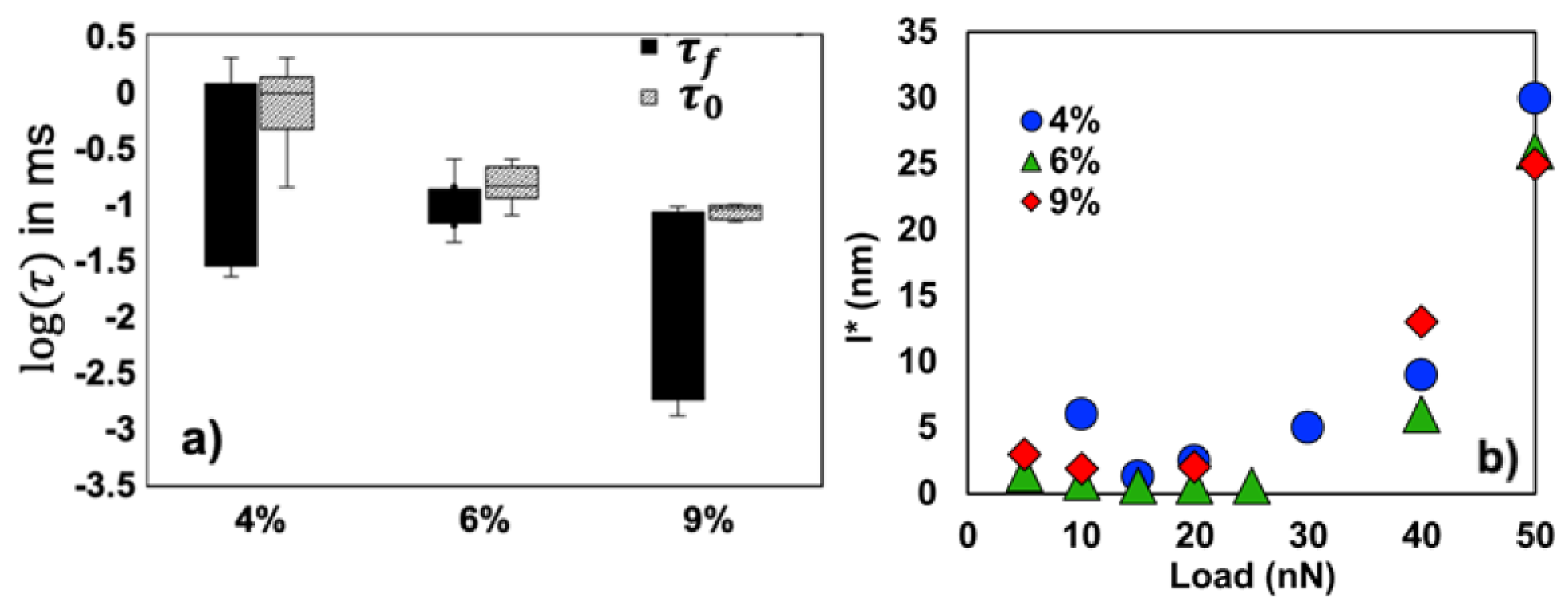
Figure 12a). Hence, the longer bond lifetime justifies the higher friction of PAAm-9% hydrogels. Our model also reflects the effect of normal load on the polymer characteristics.
Figure 12b shows how the yield length of the polymer at the interface significantly increases with load for the three hydrogels, which contributes to the enhanced energy dissipation with load. In addition, an increase in load leads to a remarkable decrease in
which implies that the formation of the adhesive bonds is facilitated with increasing load. This is likely because water is squeezed out and the polymer concentration in contact with the silica colloid increases. The change in
with load is much more subtle, as inferred from the small size of the boxes in
Figure 12a. The relaxation times of the polymer chains at the adhesive hydrogel-colloid contact are extended by several orders of magnitude compared to the unconfined polymer (
). Please note that this has been proposed for polymer friction before [
79]. This means that relaxation is slowed down and deviates from predictions of the scaling theory, likely due to the effect of confinement. This analysis thus provides insight into the relation between hydrogel microstructure and lubrication and shows that the confinement provided by the solid surface in an adhesive contact restricts the conformational entropy of the polymer and extends the relaxation time.
As shown in
Figure 11, the viscous contribution (i.e., the increase in friction with V at high velocities) was most prominent for PAAm-9% hydrogels in the range of investigated velocities. It should be noted that fits assuming a Newtonian behavior (
= 0) were possible (R
2 > 0.92), but a non-Newtonian behavior (shear thinning,
= −0.32) led to better fits, especially at the highest loads (
Table 6). Please note that the Newtonian viscosity (
) lies between 13 and 26 mPa∙s, and hence, it is higher than that of water. We associate both the increase of Newtonian viscosity and the non-Newtonian behavior in the respective models with the influence of the polymer on the viscous dissipation. For PAAm-6% hydrogels, the fits were slightly better assuming Newtonian behavior with a viscosity of 3–9 mPa∙s, which seems reasonable due to the smaller polymer concentration compared to PAAm-9% hydrogels. Our current research is dedicated to providing evidence for shear thinning and a more precise rheological model. While we show here the results estimated with
, the calculations were also carried out with
, which led to similar conclusions, although the viscosity parameter
was much higher in this case.
Friction force measurements on agarose hydrogels with a silica colloid are shown in
Figure 13a. The friction force is quasi constant at slow velocities, and it increases remarkably with velocity above ~10 μm/s. The friction force at varying loads can be fit well by
. Hence, consistent with the repulsive nature of the agarose hydrogel–colloid contact, the adhesive term (
) is zero. The fits to the experimental results assuming a Newtonian behavior (
7–9 mPa∙s,
= 0) are not satisfactory. In the range of investigated velocities, good fits are obtained by assuming a shear-thinning behavior, with an exponent
~ −0.3. These results need to be considered with caution because the range of velocities accessible to our AFM is limited and it is possible that the viscous dissipation deviates at higher velocities. Although the available data are not sufficient to exclude either Newtonian or non-Newtonian behavior, they are sufficient to support the influence of the polymer on the viscous dissipation. Please note that a velocity-independent term
is also needed here to reproduce the experimental results. Because the contact is repulsive, this term could be related to the hydration lubrication at slow velocities, which differs from our previous assumption [
23].
Figure 13b shows the results for a 9%-PAAm hydrogels for comparison; note the different scales on the
Y-axis of both diagrams.
In summary, the viscous-adhesive model provides a theoretical approach to quantify frictional response of hydrogels by considering an interplay of adhesive and viscous dissipation directly arising from the hydrogel’s microstructure. The model accounts for confinement effects, poroelastic deformation, and the influence of the polymer on the viscous dissipation in AFM friction force measurements. Limitations of this model and the experimental approach have also been highlighted, including the need for precisely quantifying the thickness of the sheared interfacial film, as well as elucidating the polymer’s contribution to the viscous dissipation more precisely.
7. Closing Remarks and Future Perspectives
Although the models discussed here provide key physical insights into lubrication mechanisms, the above-outlined existing knowledge is only partial and qualitative, since it is still generally not possible to quantitatively predict the frictional characteristics of hydrogels based on their microstructure and tribological conditions. One of the goals of this revision was to present the richness and complexity of the mechanisms underlying hydrogel lubrication and friction. Part of the complexity arises from the experimental challenge posed by contact with a soft, permeable material with large concentrations of water. Opportunities for future research arise from the outlined knowledge gaps, some of which are emphasized in this section.
To date, it is still unclear under which tribological conditions a fluid film effectively separates permeable surfaces, like hydrogels, yielding hydrodynamic and hydration lubrication. This is mainly due to the inherent difficulty to experimentally prove the presence of this film and to determine its thickness. Recent neutron-reflectometry measurements by Spencer’s group have demonstrated the presence of a thin film of water between hydrogels with a brush-like superficial layer and a silicon crystal upon an applied normal load [
44]. This method could be theoretically extended to examine the change of film thickness with sliding velocity under tribological conditions for both adhesive and repulsive contacts. The major limitation of this technique is, however, its accessibility, as well as its lack of fine control of the force over a wide range of values.
As described earlier, assumptions for the viscous dissipation range from hydrodynamic lubrication mediated by the Newtonian behavior of the solvent to non-Newtonian shear of the hydrogel interfacial region and a polymer-relaxation lubrication mechanism. While all of these assumptions are consistent with experimental observations, measurements dedicated to examining the interfacial rheology of adhesive and repulsive contacts with hydrogels are still needed to provide fundamental insight into the interfacial behavior. Rheological models that specifically account for the time-dependent variation of friction in static contacts have been already considered [
29,
30,
71]. The Surface Forces Apparatus (SFA) is a well-known method for precisely determining the thickness of (sub)nanometer fluid films, normal surface forces and friction. One advantage of this method is that the measurements of the film thickness are also possible during shear loading, so that it can determine the change in thickness with sliding velocity. Recent experiments on cartilage [
80,
81] have demonstrated the capability of the SFA to investigate hydrogel-like materials, specifically their response to compression and steady shear. The SFA has been also used to investigate the nanorheology of polymer films [
77,
82,
83], and we believe that extensions of the SFA could help understand the interfacial rheology of hydrogels under tribological conditions at the nanoscale, as well.
This review did not focus on the effects of chemical make-up and charge of hydrogel surfaces. Precedent works by Gong [
20] and Sokoloff [
15] have developed models for charged hydrogel friction. Between similarly charged surfaces, a fluid film can be expected [
15]. Interestingly, it has been shown that friction can be controlled by adjusting the local molecular conformation of a polyelectrolyte brush via an alternating electric field [
84]. The intensity of the applied field can regulate the stretching of the polymer chain while sliding, and thereby, the degree of interpenetration between opposite polymer brushes at the interface. The dynamics of the response is controlled by the relaxation times of the polyelectrolyte. While the molecular-level response to an electric field is relatively quick, less is known about the response dynamics of charged hydrogels. Electrotunable behavior offers opportunities for applications in soft robotics, among others, and hence, it is not only fundamentally interesting but also important for these applications. Furthermore, varying the fluid film properties through the modulation of an applied electric field or of the charge density of the hydrogel offers a new avenue to elucidate the mechanism of viscous and electroviscous dissipation.
The complexity of the lubrication mechanisms mediated by hydrogels also relies on other factors, including contact roughness and wear, not discussed in this review. Surface roughness can significantly affect the frictional characteristics. For example, while the friction at relatively smooth hydrogel surfaces follows well the elastic-hydrodynamic friction model by Gong [
25], hydrogels with a surface roughness in the microscale (1–10 μm) exhibit only a velocity-weakening regime. Similarly, the roughness of the hard surface is also shown to play a role in the frictional response below the critical velocity [
26,
85]. On the one hand, hydrogels have an inherent surface roughness owing to polymer dynamics at the interface. On the other, hydrogels with modulated surface topology can be prepared. Hence, it seems imperative to elucidate the influence of surface roughness on the lubrication mechanisms and friction models. Similarly, the relation between frictional dissipation and wear is still not well understood. While several works have examined the tribologically induced wear of hydrogels that can serve as biological replacement materials [
19,
86,
87,
88], the understanding of the mechanisms underlying tribologically promoted wear is not only lacking, but even more, the correlation is debated. Here, it is worth mentioning the work by Bonyadi et al. [
89], who performed systematic measurements to correlate wear, friction and surface stiffness of polyacrylamide hydrogels. The study revealed a temporary stiffening of the surface layer upon wear, which undergoes osmotic re-swelling, regains the pristine surface stiffness and corresponding low friction. The authors attributed this behavior to the self-regenerating compliance and surface structure of hydrogels. Although more studies are needed to understand how a self-regeneration of chemically crosslinked hydrogels is possible, these results announce a new direction of research.
Another relevant phenomenon for the frictional dissipation of soft materials is static friction. Although only a handful of studies have focused on static friction of hydrogels [
54,
90,
91,
92], there is sufficient experimental evidence demonstrating the dependence of adhesion and static friction on contact time. While static friction is out of the scope of this review, it is worth mentioning that wear of soft biological materials has often been related to adhesion and static friction [
93,
94,
95], and hence, future research should be dedicated to improving our understanding of the static friction of hydrogels, as well.
To conclude, hydrogels are undoubtedly of great interest to the tribology community, but there still remains much work to be done before we can design hydrogels for targeted tribological applications. Elucidating hydrogel lubrication mechanisms is not only paramount to understand better biolubrication, but also to advance the knowledge required to achieve this design goal.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}