2022 WUOF/SIU International Consultation on Urological Diseases: Genetics and Tumor Microenvironment of Renal Cell Carcinoma
 , , , , , and
, , , , , and 
Abstract
:Introduction
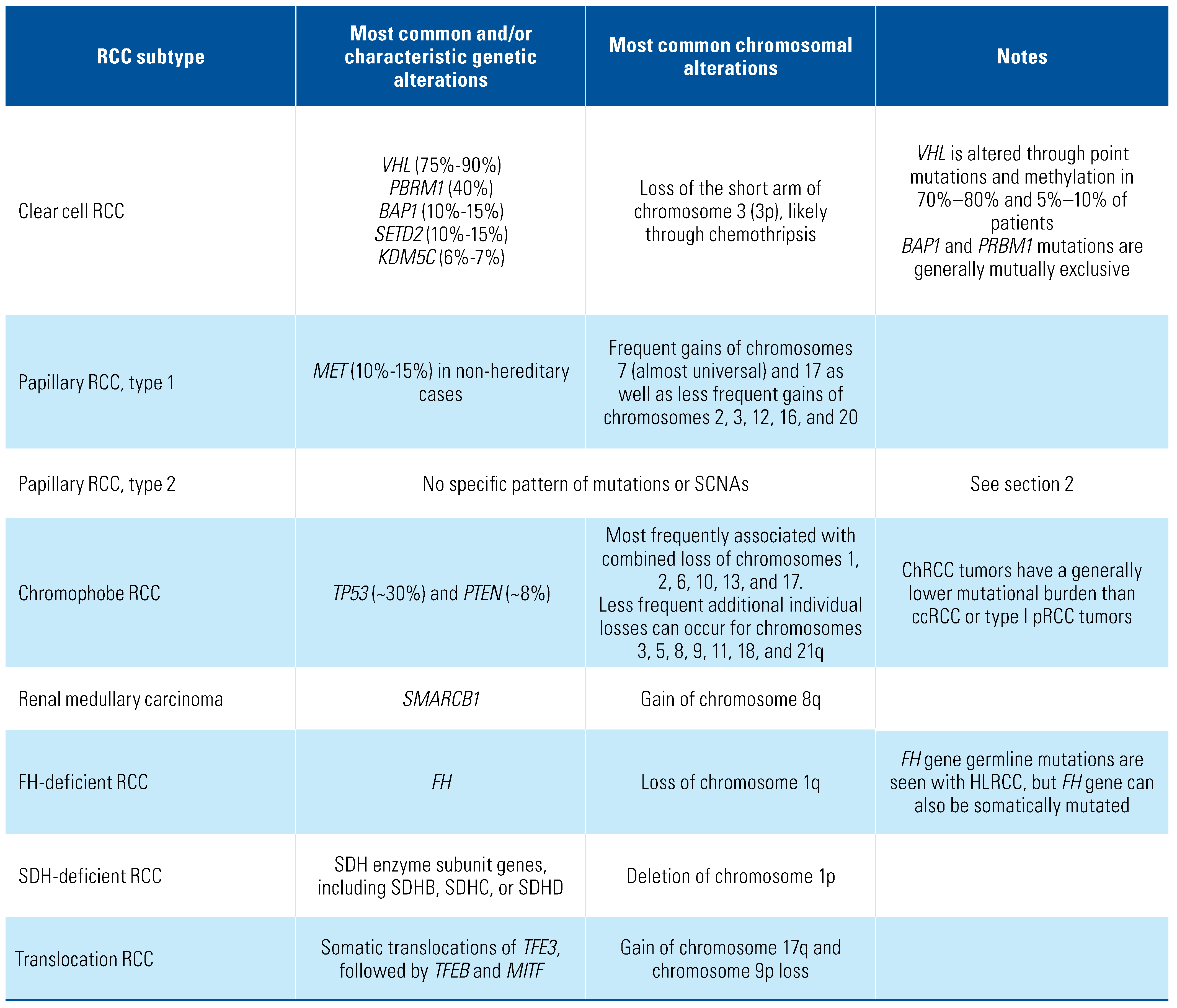
Genetics of Clear Cell RCC
Genetics of Non-Clear Cell Carcinoma
Papillary renal cell carcinoma
Chromophobe renal cell carcinoma (chRCC)
Medullary Renal Carcinoma
FH-deficient and SDH-deficient renal cell carcinoma
Translocation renal cell carcinoma involving
TFE3, TFEB, or MITF gene fusions
Tumor Microenvironment of RCC
Summary
Conflicts of Interest
Abbreviations
| ccRCC | clear cell renal cell carcinoma |
| FH | fumarate hydratase |
| RCC | renal cell carcinoma |
| SCNAs | somatic copy number alterations |
| SWI/SNF | switching defective/sucrose non-fermenting |
| TAMs | tumor-associated macrophages |
| TME | tumor microenvironment |
References
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008, 27, 5904–5912. Available at: https:// www.nature.com/articles/onc2008271. Accessed April 13, 2022. [CrossRef] [PubMed]
- Rappold, P.M.; Silagy, A.W.; Kotecha, R.R.; Hakimi, A.A. Immune checkpoint blockade in renal cell carcinoma. J Surg Oncol. 2021, 123, 739–750. Available at: https://onlinelibrary.wiley.com/doi/10.1002/jso.26339. Accessed August 12, 2021. [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013, 45, 860–867. Available at: http://www.nature.com/ articles/ng.2699. Accessed October 7, 2021. [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network TCGAR. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013, 499, 43–49. Available at: http://www.ncbi.nlm.nih.gov/ pubmed/23792563. Accessed December 24, 2021. [CrossRef] [PubMed]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.R.; O'Brien, T.; et al. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx Renal. Cell. 2018, 173, 611–623.e17. Available at: https://www.sciencedirect.com/science/article/pii/ S0092867418301648?pes=vor. Accessed March 28, 2022. [CrossRef] [PubMed]
- Brugarolas, J. Molecular genetics of clear-cell renal cell carcinoma. J Clin Oncol. 2014, 32, 1968–1976. Available at: http://ascopubs.org/doi/10.1200/JCO.2012.45.2003. Accessed December 24, 2021. [CrossRef]
- D’Avella, C.; Abbosh, P.; Pal, S.K.; Geynisman, D.M. Mutations in renal cell carcinoma. Urol Oncol Semin Orig Investig. 2020, 38, 763–773. Available at: https://www.sciencedirect.com/science/article/pii/S1078143918304368. Accessed December 24, 2021. [CrossRef]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011, 469, 539–542. Available at: http://www.nature.com/articles/nature09639. Accessed April 7, 2022. [CrossRef] [PubMed]
- Masliah-Planchon, J.; Bièche, I.; Guinebretière, J.-M.; Bourdeaut, F.; Delattre, O. SWI/SNF chromatin remodeling and human malignancies. Annu Rev Pathol. 2015, 10, 145–171. Available at: https://www.annualreviews.org/doi/10.1146/annurev-pathol-012414-040445. Accessed April 3, 2022. [CrossRef]
- Wilson, B.G.; Roberts, C.W.M. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011, 11, 481–492. Available at: http://www. nature.com/articles/nrc3068. Accessed April 7, 2022. [CrossRef]
- Peña-Llopis, S.; Vega-Rubín-de-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012, 44, 751–759. Available at: http://www.nature.com/articles/ng.2323. Accessed December 24, 2021. [CrossRef] [PubMed]
- Yu, H.; Mashtalir, N.; Daou, S.; Hammond-Martel, I.; Ross, J.; Sui, G.; et al. The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol Cell Biol. 2010, 30, 5071–5085. Available at: https://journals.asm.org/doi/10.1128/MCB.00396-10. Accessed April 7, 2022. [CrossRef] [PubMed]
- Machida, Y.J.; Machida, Y.; Vashisht, A.A.; Wohlschlegel, J.A.; Dutta, A. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J Biol Chem. 2009, 284, 34179–34188. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0021925820376997. Accessed April 24, 2022. [CrossRef]
- Peña-Llopis, S.; Christie, A.; Xie, X.-J.; Brugarolas, J. Cooperation and antagonism among cancer genes: the renal cancer paradigm. Cancer Res. 2013, 73, 4173–4179. Available at: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-13-0360. Accessed April 24, 2022. [CrossRef]
- González-Rodríguez, P.; Engskog-Vlachos, P.; Zhang, H.; Murgoci, A.-N.; Zerdes, I.; Joseph, B. SETD2 mutation in renal clear cell carcinoma suppress autophagy via regulation of ATG12. Cell Death Dis. 2020, 11, 1–15. Available at: https://doi.org/10.1038/s41419-020-2266-x. Accessed April 19, 2022. [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011, 144, 27–40. Available at: https://www.sciencedirect.com/science/article/pii/S0092867410013772. Accessed April 7, 2022. [CrossRef] [PubMed]
- Turajlic, S.; Larkin, J.; Swanton, C. SnapShot: renal cell carcinoma. Cell. 2015, 163, 1556–1556.e1. Available at: https://www.sciencedirect.com/science/article/pii/S0092867415015457?via%3Dihub. Accessed April 7, 2022. [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012, 366, 883–892. Available at: http://www.nejm.org/doi/abs/10.1056/NEJMoa1113205. Accessed April 7, 2022. [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell. 2018, 173, 581–594.e12. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx Renal. Cell. 2018, 173, 595–610.e11. Available at: https://www.sciencedirect.com/science/article/pii/S0092867418303751?pes=vor. Accessed March 28, 2022. [CrossRef]
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: linear, branching, neutral or punctuated? Biochim Biophys Acta Rev Cancer. 2017, 1867, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Mandriota, S.J.; Turner, K.J.; Davies, D.R.; Murray, P.G.; Morgan, N.V.; Sowter, H.M.; et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002, 1, 459–468. Available at: https://www.sciencedirect.com/science/article/pii/ S1535610802000715?via%3Dihub. Accessed April 7, 2022. [CrossRef] [PubMed]
- Wang, S.-S.; Gu, Y.-F.; Wolff, N.; Stafanius, K.; Christie, A.; Dey, A.; et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc Natl Acad Sci. 2014, 111, 16538–16543. Available at: https://pnas.org/doi/full/10.1073/pnas.1414789111. Accessed April 13, 2022. [CrossRef] [PubMed]
- Gu, Y.-F.; Cohn, S.; Christie, A.; McKenzie, T.; Wolff, N.; Do, Q.N.; et al. Modeling renal cell carcinoma in mice: Bap1 and Pbrm1 inactivation drive tumor grade. Cancer Discov. 2017, 7, 900–917. Available at: http://cancerdiscovery.aacrjournals.org/lookup/doi/10.1158/2159-8290.CD-17-0292. Accessed April 24, 2022. [CrossRef] [PubMed]
- Kapur, P.; Peña-Llopis, S.; Christie, A.; Zhrebker, L.; Pavía-Jiménez, A.; Rathmell, W.K.; et al. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol. 2013, 14, 159–167. Available at: https://www.sciencedirect. com/science/article/pii/S1470204512705843?via%3Dihub. Accessed December 23, 2021. [CrossRef] [PubMed]
- Singla, N.; Xie, Z.; Zhang, Z.; Gao, M.; Yousuf, Q.; Onabolu, O.; et al. Pancreatic tropism of metastatic renal cell carcinoma. JCI Insight. 2020, 5, e134564. Available at: https://insight.jci.org/articles/view/134564. Accessed April 24, 2022. [CrossRef]
- Ho, T.H.; Kapur, P.; Joseph, R.W.; Serie, D.J.; Eckel-Passow, J.E.; Tong, P.; et al. Loss of histone H3 lysine 36 trimethylation is associated with an increased risk of renal cell carcinoma-specific death. Mod Pathol. 2016, 29, 34–42. Available at: http://www.nature.com/articles/modpathol2015123. Accessed April 24, 2022. [CrossRef] [PubMed]
- Delahunt, B.; Eble, J.N. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997, 10, 537–544. Available at: http://www.ncbi.nlm.nih.gov/ pubmed/9195569. Accessed October 2, 2022. [PubMed]
- Jiang, F.; Richter, J.; Schraml, P.; Bubendorf, L.; Gasser, T.; Sauter, G.; et al. Chromosomal imbalances in papillary renal cell carcinoma: genetic differences between histological subtypes. Am J Pathol. 1998, 153, 1467–1473. [Google Scholar] [CrossRef]
- Foster, K.; Crossey, P.; Cairns, P.; Hetherington, J.W.; Richards, F.M.; Jones, M.H.; et al. Molecular genetic investigation of sporadic renal cell carcinoma: analysis of allele loss on chromosomes 3p, 5q, 11p, 17 and 22. Br J Cancer. 1994, 69, 230–234. Available at: http://www.nature.com/articles/bjc199444. Accessed April 3, 2022. [CrossRef]
- Durinck, S.; Stawiski, E.W.; Pavía-Jiménez, A.; Modrusan, Z.; Kapur, P.; Jaiswal, B.S.; et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. 2015, 47, 13–21. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016, 374, 135–145. Available at: http://www.nejm.org/doi/10.1056/NEJMoa1505917. Accessed April 3, 2022.
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, 313–326.e5. Available at: https://www.sciencedirect.com/ science/article/pii/S2211124718304364?via%3Dihub. Accessed April 3, 2022. [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.-M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto- oncogene in papillary renal carcinomas. Nat Genet. 1997, 16, 68–73. Available at: http://www.nature.com/articles/ng0597-68. Accessed April 3, 2022. [CrossRef] [PubMed]
- Chen, Y.-B.; Xu, J.; Skanderup, A.J.; Dong, Y.; Brannon, A.R.; Wang, L.; et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun. 2016, 7, 13131. Available at: http://www.nature.com/articles/ncomms13131. Accessed April 12, 2022. [CrossRef] [PubMed]
- Moch, H.; Amin, M.B.; Berney, D.M.; Compérat, E.M.; Gill, A.J.; Hartmann, A.; et al. The 2022 World Health Organization Classification of Tumours of the Urinary System and Male Genital Organs—Part A: Renal, Penile, and Testicular Tumours. Eur Urol. 2022. Available at: https://www.sciencedirect.com/science/article/pii/S0302283822024678#b0220. Accessed October 2, 2022. [CrossRef] [PubMed]
- Al-Obaidy, K.I.; Eble, J.N.; Cheng, L.; Williamson, S.R.; Sakr, W.A.; Gupta, N.; et al. Papillary renal neoplasm with reverse polarity. Am J Surg Pathol. 2019, 43, 1099–1111. Available at: https://journals.lww.com/00000478-201908000-00011. Accessed October 2, 2022. [CrossRef] [PubMed]
- Argani, P.; Reuter, V.E.; Eble, J.N.; Vlatkovic, L.; Yaskiv, O.; Swanson, D.; et al. Biphasic hyalinizing psammomatous renal cell carcinoma (BHP RCC): a distinctive neoplasm associated with somatic NF2 mutations. Am J Surg Pathol. 2020, 44, 901–916. Available at: http://www.ncbi.nlm.nih.gov/pubmed/32217839. Accessed October 2, 2022. [CrossRef] [PubMed]
- Brunelli, M.; Eble, J.N.; Zhang, S.; Martignoni, G.; Delahunt, B.; Cheng, L. Eosinophilic and classic chromophobe renal cell carcinomas have similar frequent losses of multiple chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this pattern of genetic abnormality is not present in renal oncocytoma. Mod Pathol. 2005, 18, 161–169. Available at: http://www.nature.com/articles/3800286. Accessed April 3, 2022. [CrossRef]
- Speicher, M.R.; Schoell, B.; du Manoir, S.; Schröck, E.; Ried, T.; Cremer, T.; et al. Specific loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am J Pathol. 1994, 145, 356–364. [Google Scholar]
- Ball, M.W.; Gorin, M.A.; Drake, C.G.; Hammers, H.J.; Allaf, M.E. The landscape of whole-genome alterations and pathologic features in genitourinary malignancies: an analysis of the cancer genome atlas. Eur Urol Focus. 2017, 3, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Casuscelli, J.; Weinhold, N.; Gundem, G.; Wang, L.; Zabor, E.C.; Drill, E.; et al. Genomic landscape and evolution of metastatic chromophobe renal cell carcinoma. JCI Insight. 2017, 2, e92688. Available at: https://insight.jci.org/articles/view/92688. Accessed April 12, 2022. [CrossRef] [PubMed]
- Iacovelli, R.; Modica, D.; Palazzo, A.; Trenta, P.; Piesco, G.; Cortesi, E. Clinical outcome and prognostic factors in renal medullary carcinoma: a pooled analysis from 18 years of medical literature. Can Urol Assoc J. 2015, 9, E172–E177. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, A.A.; Koi, P.T.; Milhoua, P.M.; Blitman, N.M.; Li, M.; Hugec, V.; et al. Renal medullary carcinoma: the Bronx experience. Urology. 2007, 70, 878–882. Available at: https://www.sciencedirect.com/science/article/pii/S0090429507018092?via%3Dihub. Accessed April 3, 2022. [CrossRef] [PubMed]
- Ezekian, B.; Englum, B.; Gilmore, B.F.; Nag, U.P.; Kim, J.; Leraas, H.J.; et al. Renal medullary carcinoma: a national analysis of 159 patients. Pediatr Blood Cancer. 2017, 64, e26609. Available at: https://onlinelibrary.wiley.com/doi/10.1002/pbc.26609. Accessed April 3, 2022. [CrossRef] [PubMed]
- Davis, C.J.; Mostofi, F.K.; Sesterhenn, I.A. Renal medullary carcinoma. The seventh sickle cell nephropathy. Am J Surg Pathol. 1995, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maroja Silvino, M.C.; Venchiarutti Moniz, C.M.; Munhoz Piotto, G.H.; Siqueira, S.; Galapo Kann, A.; Dzik, C. Renal medullary carcinoma response to chemotherapy: a referral center experience in Brazil. Rare Tumors. 2013, 5, e44. Available at: http://journals.sagepub.com/doi/10.4081/rt.2013.e44. Accessed April 3, 2022. [CrossRef] [PubMed]
- Marsh, A.; Golden, C.; Hoppe, C.; Quirolo, K.; Vichinsky, E. Renal medullary carcinoma in an adolescent with sickle cell anemia. Pediatr Blood Cancer. 2014, 61, 567–567. Available at: https://onlinelibrary.wiley.com/doi/10.1002/pbc.24795. Accessed April 3, 2022. [CrossRef] [PubMed]
- Berman, L.B. Sickle cell nephropathy. JAMA. 1974, 228, 1279. Available at: http://jama.jamanetwork.com/article.aspx?doi=10.1001/jama.1974.03230350051035. Accessed April 3, 2022. [CrossRef]
- Msaouel, P.; Malouf, G.G.; Su, X.; Yao, H.; Tripathi, D.N.; Soeung, M.; et al. Comprehensive molecular characterization identifies distinct genomic and immune hallmarks of renal medullary carcinoma. Cancer Cell. 2020, 37, 720–734.e13. [Google Scholar] [CrossRef]
- Kadoch, C.; Copeland, R.A.; Keilhack, H. PRC2 and SWI/SNF chromatin remodeling complexes in health and disease. Biochemistry. 2016, 55, 1600–1614. Available at: https://pubs.acs.org/doi/10.1021/acs.biochem.5b01191. Accessed April 3, 2022. [CrossRef] [PubMed]
- Launonen, V.; Vierimaa, O.; Kiuru, M.; Isola, J.; Roth, S.; Pukkala, E.; Sistonen, P.; et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001, 98, 3387–3392. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.J.; Torres-Cabala, C.; Pinto, P.; Linehan, W.M. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007, 31, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Grubb, R.L.; Franks, M.E.; Toro, J.; Middleton, L.; Choyke, L.; Fowler, S.; et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007, 177, 2074–2079; discussion 2079–2080. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Linehan, W.M. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014, 7, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Toro, J.R.; Nickerson, M.L.; Wei, M.-H.; Warren, M.B.; Glenn, G.M.; Turner, M.L.; et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003, 73, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.-H.; Toure, O.; Glenn, G.M.; Pithukpakorn, M.; Neckers, L.; Stolle, C.; et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet. 2006, 43, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Multiple-Leiomyoma-Consortium. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002, 30, 406–410. Available at: http://www.nature.com/articles/ng849z. Accessed April 3, 2022. [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000, 287, 848–851. Available at: https://www.science.org/doi/10.1126/science.287.5454.848. Accessed April 3, 2022. [CrossRef]
- Niemann, S.; Müller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000, 26, 268–270. Available at: http://www.nature.com/articles/ng1100_268. Accessed April 3, 2022. [CrossRef]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-H.; Sourbier, C.; Kovtunovych, G.; Jeong, S.Y.; Vira, M.; Ghosh, M.; et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. 2011, 20, 315–327. Available at: https://www.sciencedirect.com/science/article/pii/S1535610811002686?via%3Dihub. Accessed April 3, 2022. [CrossRef] [PubMed]
- Yang, Y.; Lane, A.N.; Ricketts, C.J.; Sourbier, C.; Wei, M.-H.; Shuch, B.; et al. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS One. 2013, 8, e72179. Available at: https://dx.plos.org/10.1371/journal.pone.0072179. Accessed April 3, 2022. [CrossRef] [PubMed]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, M.C.; et al. Accumulation of Krebs cycle intermediates and over- expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005, 14, 2231–2239. Available at: http://academic.oup.com/hmg/article/14/15/2231/551734/Accumulation-of-Krebs-cycle- intermediates-and. Accessed April 3, 2022. [CrossRef] [PubMed]
- Saxena, N.; Maio, N.; Crooks, D.R.; Ricketts, C.J.; Yang, Y.; Wei, M.-H.; et al. SDHB-deficient cancers: the role of mutations that impair iron sulfur cluster delivery. J Natl Cancer Inst. 2016, 108, djv287. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; et al. Inhibition of α-KG- dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.-L.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005, 8, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Bardella, C.; El-Bahrawy, M.; Frizzell, N.; Adam, J.; Ternette, N.; Hatipoglu, E.; et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol. 2011, 225, 4–11. Available at: https:// onlinelibrary.wiley.com/doi/10.1002/path.2932. Accessed April 3, 2022. [CrossRef] [PubMed]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011, 20, 524–537. Available at: https://www.sciencedirect.com/science/article/pii/S1535610811003540?via%3Dihub. Accessed April 3, 2022. [CrossRef]
- Yoo, A.; Tang, C.; Zucker, M.; Fitzgerald, K.; DiNatale, R.G.; Rappold, P.M.; et al. Genomic and metabolic hallmarks of SDH- and FH-deficient renal cell carcinomas. Eur Urol Focus. 2022. Online ahead of print. Available at: https://www.eu-focus.europeanurology.com/article/S2405-4569(21)00312-6/fulltext. Accessed April 12, 2022. [CrossRef]
- Argani, P. MiT family translocation renal cell carcinoma. Semin Diagn Pathol. 2015, 32, 103–113. Available at: https://www.sciencedirect.com/science/article/pii/S0740257015000040?via%3Dihub. Accessed April 3, 2022. [CrossRef]
- Kauffman, E.C.; Ricketts, C.J.; Rais-Bahrami, S.; Yang, Y.; Merino, M.J.; Bottaro, D.P.; et al. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol. 2014, 11, 465–475. Available at: http://www.nature.com/articles/nrurol.2014.162. Accessed April 3, 2022. [CrossRef]
- La Spina, M.; Contreras, P.S.; Rissone, A.; Meena, N.K.; Jeong, E.; Martina, J.A. MiT/TFE family of transcription factors: an evolutionary perspective. Front Cell Dev Biol. 2021, 8, 1580. Available at: https://www.frontiersin.org/articles/10.3389/fcell.2020.609683/full. Accessed April 3, 2022. [CrossRef]
- Chen, F.; Zhang, Y.; Şenbabaoğlu, Y.; Ciriello, G.; Yang, L.; Reznik, E.; et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep. 2016, 14, 2476–2489. [Google Scholar] [CrossRef]
- Argani, P.; Reuter, V.E.; Zhang, L.; Sung, Y.S.; Ning, Y.; Epstein, J.I.; et al. TFEB-amplified renal cell carcinomas: an aggressive molecular subset demonstrating variable melanocytic marker expression and morphologic heterogeneity. Am J Surg Pathol. 2016, 40, 1484–1495. [Google Scholar] [CrossRef]
- Xia, Q.-Y.; Wang, X.-T.; Ye, S.-B.; Wang, X.; Li, R.; Shi, S.-S.; et al. Novel gene fusion of PRCC-MITF defines a new member of MiT family translocation renal cell carcinoma: clinicopathological analysis and detection of the gene fusion by RNA sequencing and FISH. Histopathology. 2018, 72, 786–794. Available at: https://onlinelibrary.wiley.com/doi/10.1111/his.13439. Accessed April 3, 2022. [CrossRef]
- Şenbabaoğlu, Y.; Gejman, R.S.; Winer, A.G.; Liu, M.; Van Allen, E.M.; de Valasco, G.; et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016, 17, 231. Available at: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-1092-z. Accessed August 12, 2021. [CrossRef] [PubMed]
- Wang, T.; Lu, R.; Kapur, P.; Jaiswal, B.S.; Hannan, R.; Zhang, Z.; et al. An empirical approach leveraging tumorgrafts to dissect the tumor microenvironment in renal cell carcinoma identifies missing link to prognostic inflammatory factors. Cancer Discov. 2018, 8, 1142–1155. Available at: http://cancerdiscovery.aacrjournals.org/lookup/doi/10.1158/2159-8290.CD-17-1246. Accessed April 24, 2022. [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat Immunol. 2011, 12, 492–499. Available at: http://www.nature.com/articles/ni.2035. Accessed April 13, 2022. [CrossRef] [PubMed]
- Speiser, D.E.; Ho, P.-C.; Verdeil, G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016, 16, 599–611. Available at: http://www. nature.com/articles/nri.2016.80. Accessed April 13, 2022. [CrossRef]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell. 2021, 39, 662–677. Available at: https://www.sciencedirect.com/science/article/pii/S1535610821001653?via%3Dihub. Accessed August 12, 2021. [CrossRef]
- Vuong, L.; Kotecha, R.R.; Voss, M.H.; Hakimi, A.A. Tumor microenvironment dynamics in clear- cell renal cell carcinoma. Cancer Discov. 2019, 9, 1349–1357. Available at: www.aacrjournals.org. Accessed September 14, 2021. [CrossRef]
- Braun, D.A.; Street, K.; Burke, K.P.; Cookmeyer, D.L.; eDenize, T.; Pederson, C.B.; et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell. 2021, 39, 632–648.e8. Available at: https://www.sciencedirect.com/science/article/pii/S153561082100115X. Accessed April 12, 2022. [CrossRef] [PubMed]
- Bi, K.; He, M.X.; Bakouny, Z.; Kanodia, A.; Napolitano, S.; Wu, J.; et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell. 2021, 39, 649–661.e5. Available at: https://www.sciencedirect.com/science/article/pii/ S1535610821001173?via%3Dihub. Accessed April 12, 2022. [CrossRef]
- Chakiryan, N.H.; Kimmel, G.J.; Kim, Y.; Hajiran, A.; Aydin, A.M.; Zemp, L.; et al. Spatial clustering of CD68+ tumor associated macrophages with tumor cells is associated with worse overall survival in metastatic clear cell renal cell carcinoma. PLoS One. 2021, 16, e0245415. Available at: https://dx.plos.org/10.1371/journal.pone.0245415. Accessed April 12, 2022. [CrossRef]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; et al. An immune atlas of clear cell renal cell carcinoma. Cell. 2017, 169, 736–749. Available at: https://www.sciencedirect.com/science/article/pii/S0092867417304294. Accessed April 12, 2022. [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Wladner, M.; Obenauf, A.C.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013, 39, 782–795. Available at: https://www.sciencedirect.com/science/article/pii/S1074761313004378?via%3Dihub. Accessed August 12, 2021. [CrossRef]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018, 24, 749–757. Available at: http://www.nature.com/articles/s41591-018-0053-3. Accessed September 12, 2021. [CrossRef]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Sant' Angleo, M.; Forman, J.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat Med. 2020, 26, 909–918. Available at: http://www.nature.com/articles/s41591-020-0839-y. Accessed December 16, 2021. [CrossRef]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019, 576, 465–470. Available at: http://www.nature.com/articles/s41586-019-1836-5. Accessed April 12, 2022. [CrossRef] [PubMed]
- Hakimi, A.A.; Voss, M.H.; Kuo, F.; Sanchez, A.; Liu, M.; Nixon, B.G.; et al. Transcriptomic profiling of the tumor microenvironment reveals distinct subgroups of clear cell renal cell cancer: data from a randomized phase III trial. Cancer Discov. 2019, 9, 510–525. Available at: http://cancerdiscovery.aacrjournals.org/lookup/doi/10.1158/2159-8290.CD-18-0957. Accessed August 12, 2021. [CrossRef] [PubMed]
- Motzer, R.J.; Choueiri, T.K.; McDermott, D.F.; Powles, T.; Yao, J.; Ammar, R.; et al. Biomarker analyses from the phase III CheckMate 214 trial of nivolumab plus ipilimumab (N+I) or sunitinib (S) in advanced renal cell carcinoma (aRCC). J Clin Oncol. 2020, 38, 5009–5009. Available at: https://ascopubs.org/doi/10.1200/JCO.2020.38.15_suppl.5009. Accessed December 21, 2021. [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet. 2019, 393, 2404–2415. Available at: https://www.sciencedirect.com/science/article/pii/ S0140673619307238?via%3Dihub. Accessed December 17, 2021. [CrossRef] [PubMed]
- Beuselinck, B.; Job, S.; Becht, E.; Karadimou, A.; Verkarre, V.; Couchy, G.; et al. Molecular subtypes of clear cell renal cell carcinoma are associated with sunitinib response in the metastatic setting. Clin Cancer Res. 2015, 21, 1329–1339. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25583177. Accessed January 17, 2022. [CrossRef] [PubMed]
- Verbiest, A.; Couchy, G.; Job, S.; Caruana, L.; Lerut, E.; Oyen, R.; et al. Molecular subtypes of clear-cell renal cell carcinoma are prognostic for outcome after complete metastasectomy. Eur Urol. 2018, 74, 474–480. Available at: https://www.sciencedirect.com/science/article/pii/S0302283818300976. Accessed October 2, 2022. [CrossRef]
- Beuselinck, B.; Verbiest, A.; Couchy, G.; Job, S.; de Reynies, A.; Meiller, C.; et al. Pro-angiogenic gene expression is associated with better outcome on sunitinib in metastatic clear-cell renal cell carcinoma. Acta Oncol. 2018, 57, 498–508. Available at: https://www.tandfonline.com/doi/full/10.1080/0284186X.2017.1388927. Accessed January 17, 2022. [CrossRef]
- Vano, Y.-A.; Elaidi, R.; Bennamoun, M.; Chevreau, C.; Borchiellini, D.; Pannier, D.; et al. Nivolumab, nivolumab–ipilimumab, and VEGFR- tyrosine kinase inhibitors as first-line treatment for metastatic clear-cell renal cell carcinoma (BIONIKK): a biomarker-driven, open-label, non-comparative, randomised, phase 2 trial. Lancet Oncol. 2022, 23, 612–624. Available at: https://www.sciencedirect.com/science/article/pii/S1470204522001280#bib6. Accessed October 2, 2022. [CrossRef]
- Koh, M.Y.; Sayegh, N.; Agarwal, N. Seeing the forest for the trees—single- cell atlases link CD8+ T cells and macrophages to disease progression and treatment response in kidney cancer. Cancer Cell. 2021, 39, 594–596. Available at: https://www.sciencedirect.com/science/article/pii/S1535610821001665?via%3Dihub. Accessed April 12, 2022. [CrossRef]

 |
This is an open access article under the terms of a license that permits non-commercial use, provided the original work is properly cited. © 2022 The Authors. Société Internationale d'Urologie Journal, published by the Société Internationale d'Urologie, Canada.
Share and Cite
Khaleel, S.; Ricketts, C.; Linehan, W.M.; Ball, M.; Manley, B.; Turajilic, S.; Brugarolas, J.; Hakimi, A. 2022 WUOF/SIU International Consultation on Urological Diseases: Genetics and Tumor Microenvironment of Renal Cell Carcinoma. Soc. Int. Urol. J. 2022, 3, 386-396. https://doi.org/10.48083/BLPV3411
Khaleel S, Ricketts C, Linehan WM, Ball M, Manley B, Turajilic S, Brugarolas J, Hakimi A. 2022 WUOF/SIU International Consultation on Urological Diseases: Genetics and Tumor Microenvironment of Renal Cell Carcinoma. Société Internationale d’Urologie Journal. 2022; 3(6):386-396. https://doi.org/10.48083/BLPV3411
Chicago/Turabian StyleKhaleel, Sari, Christopher Ricketts, W. Marston Linehan, Mark Ball, Brandon Manley, Samra Turajilic, James Brugarolas, and Ari Hakimi. 2022. "2022 WUOF/SIU International Consultation on Urological Diseases: Genetics and Tumor Microenvironment of Renal Cell Carcinoma" Société Internationale d’Urologie Journal 3, no. 6: 386-396. https://doi.org/10.48083/BLPV3411
APA StyleKhaleel, S., Ricketts, C., Linehan, W. M., Ball, M., Manley, B., Turajilic, S., Brugarolas, J., & Hakimi, A. (2022). 2022 WUOF/SIU International Consultation on Urological Diseases: Genetics and Tumor Microenvironment of Renal Cell Carcinoma. Société Internationale d’Urologie Journal, 3(6), 386-396. https://doi.org/10.48083/BLPV3411