2022 WUOF/SIU International Consultation on Urological Diseases: Hereditary Renal Cell Carcinoma Syndromes

{kind=link}
{kind=link}
Abstract
:Introduction
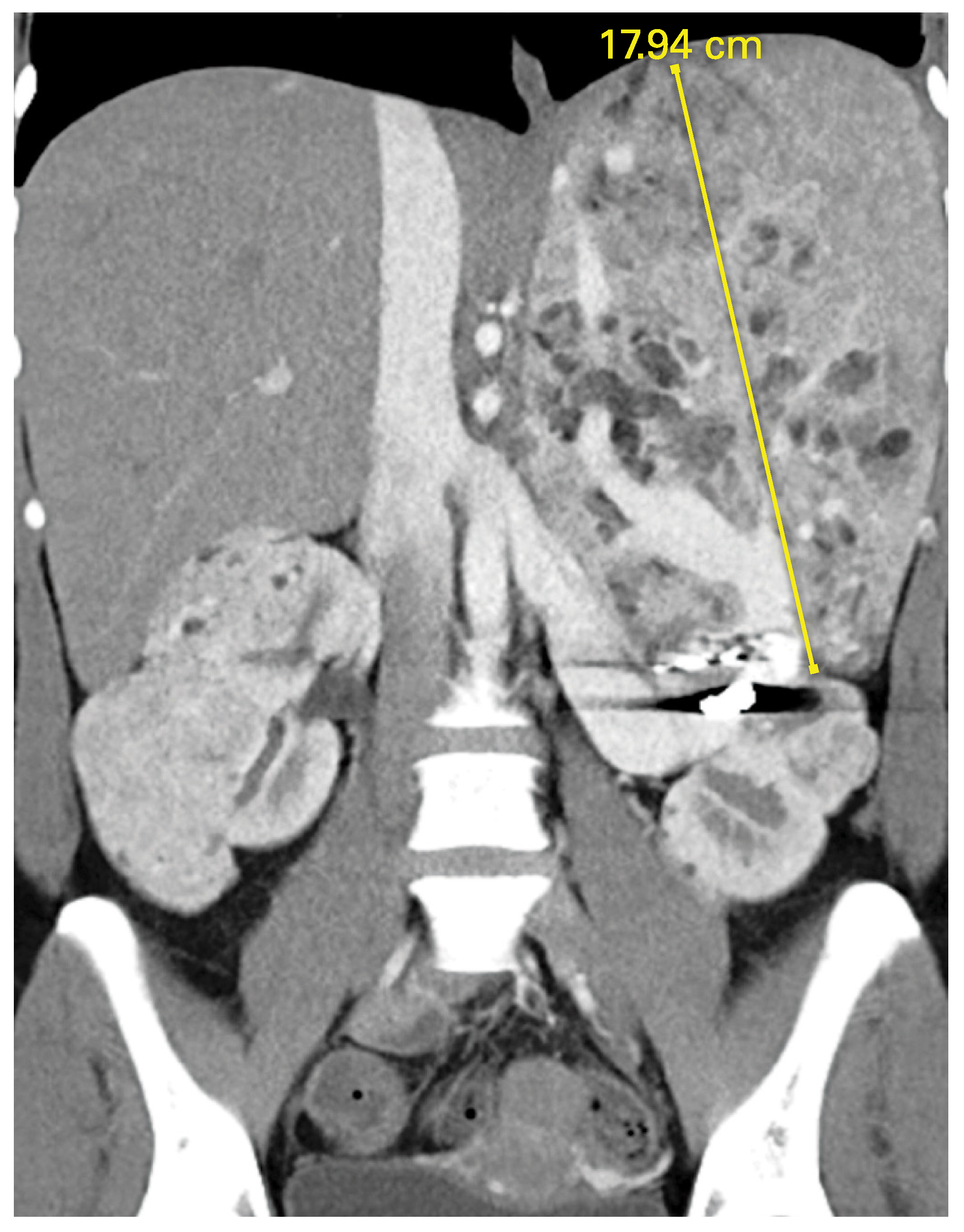
von Hippel-Lindau Disease (VHL)
Birt-Hogg-Dubé Syndrome (BHD)
Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC)
Hereditary Papillary Renal Cell Carcinoma (HPRC)
Tuberous Sclerosis Complex (TSC)
PTEN hamartoma tumor syndrome (PHTS)
BAP1 Tumor Predisposition Syndrome (BAP1-TPDS)
Succinate Dehydrogenase (SDH)-Deficient RCC
Hereditary Hyperparathyroidism Jaw Tumor Syndrome (HPT-JT)
Conclusion
Conflicts of Interest
Abbreviations
| AML | angiomyolipoma |
| BAP1-TPDS BAP1 | tumor predisposition syndrome |
| BHD | Birt-Hogg-Dubé syndrome |
| ccRCC | clear cell renal cell carcinoma |
| CNS | central nervous system |
| CS | Cowden syndrome |
| CT | computed tomography |
| ESC RCC | eosinophilic solid cystic RCC |
| FH | fumarate hydratase |
| HLRCC | hereditary leiomyomatosis and renal cell carcinoma |
| MRI | magnetic resonance imaging |
| mTOR | mammalian target of rapamycin |
| PHTS PTEN | hamartoma tumor syndrome |
| RCC | renal cell carcinoma |
| TSC | tuberous sclerosis complex |
| TSG | tumor suppressor gene |
| VHL | von Hippel-Lindau disease |
References
- Shuch, B.; Vourganti, S.; Ricketts, C.J.; Middleton, L.; Peterson, J.; Merino, M.J.; et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol. 2014, 32, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Gomella, P.T.; Linehan, W.M.; Ball, M.W. Precision Surgery and Kidney Cancer: Knowledge of Genetic Alterations Influences Surgical Management. Genes (Basel). 2021, 12. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; et al. The metabolic basis of kidney cancer. Cancer Discov. 2019, 9, 1006–1021. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; et al. von Hippel-Lindau disease. Lancet. 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Duffey, B.G.; Choyke, P.L.; Glenn, G.; Grubb, R.L.; Venzon, D.; Linehan, W.M.; et al. The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease. J Urol. 2004, 172, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.M.; Choyke, P.L.; Glenn, G.; Lyne, J.C.; Rayford, W.; Venzon, D.; et al. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol. 1999, 161, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.M.; Choyke, P.L.; Weiss, G.; Manolatos, C.; Long, J.; Reiter, R.; et al. Parenchymal sparing surgery in patients with hereditary renal cell carcinoma. J Urol. 1995, 153 (3 Pt 2), 913–916. [Google Scholar]
- Herring, J.C.; Enquist, E.G.; Chernoff, A.; Linehan, W.M.; Choyke, P.L.; Walther, M.M. Parenchymal sparing surgery in patients with hereditary renal cell carcinoma: 10-year experience. J Urol. 2001, 165, 777–781. [Google Scholar] [CrossRef]
- Steinbach, F.; Novick, A.C.; Zincke, H.; Miller, D.P.; Williams, R.D.; Lund, G.; et al. Treatment of renal cell carcinoma in von Hippel-Lindau disease: a multicenter study. J Urol. 1995, 153, 1812–1816. [Google Scholar] [CrossRef]
- Johnson, A.; Sudarshan, S.; Liu, J.; Linehan, W.M.; Pinto, P.A.; Bratslavsky, G. Feasibility and outcomes of repeat partial nephrectomy. J Urol. 2008, 180, 89–93, discussion. [Google Scholar] [CrossRef] [PubMed]
- Bratslavsky, G.; Liu, J.J.; Johnson, A.D.; Sudarshan, S.; Choyke, P.L.; Linehan, W.M.; et al. Salvage partial nephrectomy for hereditary renal cancer: feasibility and outcomes. J Urol. 2008, 179, 67–70. [Google Scholar] [CrossRef]
- Carrion, D.M.; Linares-Espinos, E.; Rios Gonzalez, E.; Bazan, A.A.; Alvarez-Maestro, M.; Martinez-Pineiro, L. Invasive management of renal cell carcinoma in von Hippel-Lindau disease. Cent European J Urol. 2020, 73, 167–172. [Google Scholar]
- Park, B.K.; Kim, C.K.; Park, S.Y.; Shen, S.H. Percutaneous radiofrequency ablation of renal cell carcinomas in patients with von Hippel Lindau disease: indications, techniques, complications, and outcomes. Acta Radiol. 2013, 54, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel- Lindau Disease. N Engl J Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Birt, A.R.; Hogg, G.R.; Dube, W.J. Hereditar y multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977, 113, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Menko, F.H.; Maher, E.R.; Schmidt, L.S.; Middelton, L.A.; Aittomaki, K.; Tomlinson, I.; et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014, 13, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Linehan, W.M. Molecular genetics and clinical features of Birt-Hogg-Dube syndrome. Nat Rev Urol. 2015, 12, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Lim, D.; Pharoah, P.D.; Maher, E.R.; Marciniak, S.J. A systematic review assessing the existence of pneumothorax-only variants of FLCN. Implications for lifelong surveillance of renal tumours. Eur J Hum Genet. 2021, 29, 1595–1600. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Warren, M.B.; Toro, J.R.; Matrosova, V.; Glenn, G.; Turner, M.L.; et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002, 2, 157–164. [Google Scholar] [CrossRef]
- Houweling, A.C.; Gijezen, L.M.; Jonker, M.A.; van Doorn, M.B.; Oldenburg, R.A.; van Spaendonck-Zwarts, K.Y.; et al. Renal cancer and pneumothorax risk in Birt-Hogg-Dube syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br J Cancer. 2011, 105, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Benusiglio, P.R.; Giraud, S.; Deveaux, S.; Mejean, A.; Correas, J.M.; Joly, D.; et al. Renal cell tumour characteristics in patients with the Birt-Hogg- Dube cancer susceptibility syndrome: a retrospective, multicentre study. Orphanet J Rare Dis. 2014, 9, 163. [Google Scholar] [CrossRef]
- Stamatakis, L.; Metwalli, A.R.; Middelton, L.A.; Marston Linehan, W. Diagnosis and management of BHD-associated kidney cancer. Fam Cancer. 2013, 12, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Sattler, E.C.; Syunyaeva, Z.; Reithmair, M.; Dempke, W.; Steinlein, O.K. Colorectal cancer risk in families with Birt-Hogg-Dube syndrome increased. Eur J Cancer. 2021, 151, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Furihata, M.; Hong, S.B.; Tessarollo, L.; Haines, D.C.; Southon, E.; et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. 2008, 100, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Gijezen, L.M.; Vernooij, M.; Martens, H.; Oduber, C.E.; Henquet, C.J.; Starink, T.M.; et al. Topical rapamycin as a treatment for fibrofolliculomas in Birt- Hogg-Dube syndrome: a double-blind placebo-controlled randomized split-face trial. PLoS One. 2014, 9, e99071. [Google Scholar] [CrossRef]
- Reed, W.B.; Walker, R.; Horowitz, R. Cutaneous leiomyomata with uterine leiomyomata. Acta Derm Venereol. 1973, 53, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Launonen, V.; Vierimaa, O.; Kiuru, M.; Isola, J.; Roth, S.; Pukkala, E.; et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001, 98, 3387–3392. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002, 30, 406–410. [Google Scholar]
- Shuch, B.; Li, S.; Risch, H.; Bindra, R.S.; McGillivray, P.D.; Gerstein, M. Estimation of the carrier frequency of fumarate hydratase alterations and implications for kidney cancer risk in hereditary leiomyomatosis and renal cancer. Cancer. 2020, 126, 3657–3666. [Google Scholar] [CrossRef]
- Forde, C.; Lim, D.H.K.; Alwan, Y.; Burghel, G.; Butland, L.; Cleaver, R.; et al. Hereditary Leiomyomatosis and Renal Cell Cancer: Clinical, Molecular, and Screening Features in a Cohort of 185 Affected Individuals. Eur Urol Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Ferlicot, S.; Guillaud-Bataille, M.; Le Teuff, G.; Genestie, C.; Deveaux, S.; et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin Genet. 2017, 92, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Grubb, R.L., 3rd; Franks, M.E.; Toro, J.; Middelton, L.; Choyke, L.; Fowler, S.; et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007, 177, 2074–2079; discussion 9–80. [Google Scholar] [CrossRef]
- Chen, Y.B.; Brannon, A.R.; Toubaji, A.; Dudas, M.E.; Won, H.H.; Al-Ahmadie, H.A.; et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014, 38, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.J.; Torres-Cabala, C.; Pinto, P.; Linehan, W.M. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007, 31, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Bratslavsky, G.; Mendhiratta, N.; Daneshvar, M.; Brugarolas, J.; Ball, M.W.; Metwalli, A.; et al. Genetic risk assessment for hereditary renal cell carcinoma: Clinical consensus statement. Cancer. 2021, 127, 3957–3966. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.M.; Solomon, D.A.; Frizzell, N.; Rabban, J.T.; Zaloudek, C.; Garg, K. Morphology and Immunohistochemistry for 2SC and FH aid in detection of fumarate hydratase gene aberrations in uterine leiomyomas from young patients. Am J Surg Pathol. 2015, 39, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Kopp, R.P.; Stratton, K.L.; Glogowski, E.; Schrader, K.A.; Rau-Murthy, R.; Russo, P.; et al. Utility of prospective pathologic evaluation to inform clinical genetic testing for hereditary leiomyomatosis and renal cell carcinoma. Cancer. 2017, 123, 2452–2458. [Google Scholar] [CrossRef]
- Yamasaki, T.; Tran, T.A.; Oz, O.K.; Raj, G.V.; Schwarz, R.E.; Deberardinis, R.J.; et al. Exploring a glycolytic inhibitor for the treatment of an FH-deficient type-2 papillary RCC. Nat Rev Urol. 2011, 8, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Nikolovski, I.; Carlo, M.I.; Chen, Y.B.; Vargas, H.A. Imaging features of fumarate hydratase-deficient renal cell carcinomas: a retrospective study. Cancer Imaging. 2021, 21, 24. [Google Scholar] [CrossRef]
- Srinivasan, R.; Gurram, S.; Al Harthy, M.A.; Singer, E.A.; Sidana, A.; Shuch, B.; et al. Results from a phase II study of bevacizumab and erlotinib in subjects with advanced hereditary leiomyomatosis and renal cell cancer (HLRCC) or sporadic papillary renal cell cancer. J Clin Oncol. 2020, 38, 5004. [Google Scholar] [CrossRef]
- Carril-Ajuria, L.; Colomba, E.; Cerbone, L.; Romero-Ferreiro, C.; Crouzet, L.; Laguerre, B.; et al. Response to systemic therapy in fumarate hydratase- deficient renal cell carcinoma. Eur J Cancer. 2021, 151, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, J.P.; Nikolovski, I.; Dinatale, R.; Zucker, M.; Knezevic, A.; Patil, S.; et al. Comprehensive Molecular Characterization and Response to Therapy in Fumarate Hydratase-Deficient Renal Cell Carcinoma. Clin Cancer Res. 2021, 27, 2910–2919. [Google Scholar] [CrossRef]
- Iribe, Y.; Furuya, M.; Shibata, Y.; Yasui, M.; Funahashi, M.; Ota, J.; et al. Complete response of hereditary leiomyomatosis and renal cell cancer (HLRCC)-associated renal cell carcinoma to nivolumab and ipilimumab combination immunotherapy by: a case report. Fam Cancer. 2021, 20, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet 2018, 50, 1086–1092. [Google Scholar] [CrossRef]
- Zbar, B.; Tory, K.; Merino, M.; Schmidt, L.; Glenn, G.; Choyke, P.; et al. Hereditary papillary renal cell carcinoma. J Urol. 1994, 151, 561–566. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto- oncogene in papillary renal carcinomas. Nat Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Ferlicot, S.; Just, P.A.; Comperat, E.; Rouleau, E.; Tissier, F.; Vaessen, C.; et al. Clear cell and papillary renal cell carcinomas in hereditary papillary renal cell carcinoma (HPRCC) syndrome: a case report. Diagn Pathol. 2021, 16, 107. [Google Scholar] [CrossRef]
- Choyke, P.L.; Glenn, G.M.; Walther, M.M.; Zbar, B.; Linehan, W.M. Hereditary renal cancers. Radiology. 2003, 226, 33–46. [Google Scholar] [CrossRef]
- Lubensky, I.A.; Schmidt, L.; Zhuang, Z.; Weirich, G.; Pack, S.; Zambrano, N.; et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999, 155, 517–526. [Google Scholar] [CrossRef]
- PCGE, B. Hereditary Papillary Renal Carcinoma (PDQ®)2021.
- Choueiri, T.K.; Vaishampayan, U.; Rosenberg, J.E.; Logan, T.F.; Harzstark, A.L.; Bukowski, R.M.; et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013, 31, 181–186. [Google Scholar] [CrossRef]
- Duh, F.M.; Scherer, S.W.; Tsui, L.C.; Lerman, M.I.; Zbar, B.; Schmidt, L. Gene structure of the human MET proto-oncogene. Oncogene. 1997, 15, 1583–1586. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Kaul, K.; Braun, M.J.; Gonda, M.A.; Vande Woude, G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci U S A. 1987, 84, 6379–6383. [Google Scholar] [CrossRef]
- Carlo, M.I.; Hakimi, A.A.; Stewart, G.D.; Bratslavsky, G.; Brugarolas, J.; Chen, Y.B.; et al. Familial kidney cancer: implications of new syndromes and molecular insights. Eur Urol. 2019, 76, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, M.; Kujala, M.; Aittomaki, K. Inherited forms of renal cell carcinoma. Scand J Surg. 2004, 93, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Fadahunsi, A.T.; Sanford, T.; Linehan, W.M.; Pinto, P.A.; Bratslavsky, G. Feasibility and outcomes of partial nephrectomy for resection of at least 20 tumors in a single renal unit. J Urol. 2011, 185, 49–53. [Google Scholar] [CrossRef]
- Martinez Chanza, N.; Xie, W.; Asim Bilen, M.; Dzimitrowicz, H.; Burkart, J.; Geynisman, D.M.; et al. Cabozantinib in advanced non-clear-cell renal cell carcinoma: a multicentre, retrospective, cohort study. Lancet Oncol. 2019, 20, 581. [Google Scholar] [CrossRef]
- Osborne, J.P.; Jones, A.C.; Burley, M.W.; Jeganathan, D.; Young, J.; O'Callaghan, F.J.; et al. Non-penetrance in tuberous sclerosis. Lancet. 2000, 355, 1698. [Google Scholar] [CrossRef]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet. 2008, 372, 657–668. [Google Scholar] [CrossRef]
- Yang, P.; Cornejo, K.M.; Sadow, P.M.; Cheng, L.; Wang, M.; Xiao, Y.; et al. Renal cell carcinoma in tuberous sclerosis complex. Am J Surg Pathol. 2014, 38, 895–909. [Google Scholar] [CrossRef]
- Verhoef, S.; Bakker, L.; Tempelaars, A.M.; Hesseling-Janssen, A.L.; Mazurczak, T.; Jozwiak, S.; et al. High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet. 1999, 64, 1632–1637. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N Engl J Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Sancak, O.; Nellist, M.; Goedbloed, M.; Elfferich, P.; Wouters, C.; Maat- Kievit, A.; et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype--phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005, 13, 731–741. [Google Scholar] [CrossRef]
- Kingswood, J.C.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; et al. Renal angiomyolipoma in patients with tuberous sclerosis complex: findings from the TuberOus SClerosis registry to increase disease Awareness. Nephrol Dial Transplant. 2019, 34, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Northrup, H.; Aronow, M.E.; Bebin, E.M.; Bissler, J.; Darling, T.N.; de Vries, P.J.; et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr Neurol. 2021, 123, 50–66. [Google Scholar] [CrossRef]
- Guo, G.; Gu, L.; Zhang, X. Everolimus in invasive malignant renal epithelioid angiomyolipoma. Front Oncol. 2020, 10, 610858. [Google Scholar] [CrossRef]
- Guo, J.; Tretiakova, M.S.; Troxell, M.L.; Osunkoya, A.O.; Fadare, O.; Sangoi, A.R.; et al. Tuberous sclerosis-associated renal cell carcinoma: a clinicopathologic study of 57 separate carcinomas in 18 patients. Am J Surg Pathol. 2014, 38, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Duffy, K.; Al-Saleem, T.; Karbowniczek, M.; Ewalt, D.; Prowse, A.H.; Henske, E.P. Mutational analysis of the von hippel lindau gene in clear cell renal carcinomas from tuberous sclerosis complex patients. Mod Pathol. 2002, 15, 205–210. [Google Scholar] [CrossRef]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Schuurs-Hoeijmakers, J.H.M.; Vos, J.R. A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin Genet. 2021, 99, 219–225. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Mester, J.L.; Zhou, M.; Prescott, N.; Eng, C. Papillary renal cell carcinoma is associated with PTEN hamartoma tumor syndrome. Urology. 2012, 79, 1187–e1. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Ricketts, C.J.; Vocke, C.D.; Komiya, T.; Middelton, L.A.; Kauffman, E.C.; et al. Germline PTEN mutation Cowden syndrome: an underappreciated form of hereditary kidney cancer. J Urol. 2013, 190, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Eng CPTENHamartoma Tumor Syndrome In: Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; et al., eds. GeneReviews((R)). Seattle (WA)1993.
- Ngeow, J.; Liu, C.; Zhou, K.; Frick, K.D.; Matchar, D.B.; Eng, C. Detecting Germline PTEN Mutations among at-risk patients with cancer: an age- and sex-specific cost-effectiveness analysis. J Clin Oncol. 2015, 33, 2537–2544. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Colas, C.; Pouwels, S.; Hoogerbrugge, N.; Group, P.G.D.; European Reference Network, G. Cancer Surveillance Guideline for individuals with PTEN hamartoma tumour syndrome. Eur J Hum Genet. 2020, 28, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Wang, X.; Evans, A.J.; Campbell, S.C.; Nguyen, J.K.; Farncombe, K.M.; et al. Early-onset renal cell carcinoma in PTEN harmatoma tumour syndrome. NPJ Genom Med. 2020, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Liao, A.; Leng, N.; Pavia-Jimenez, A.; Wang, S.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Brugarolas, J. PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer J. 2013, 19, 324–332. [Google Scholar] [CrossRef]
- Shao, Y.F.; DeBenedictis, M.; Yeaney, G.; Singh, A.D. Germ Line BAP1 Mutation in Patients with Uveal Melanoma and Renal Cell Carcinoma. Ocul Oncol Pathol. 2021, 7, 340–345. [Google Scholar] [CrossRef]
- Pilarski, R.; Carlo, M.; Cebulla, C.; Abdel-Rahman, M. BAP1 Tumor predisposition syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al., editors. GeneReviews((R)). Seattle (WA)1993.
- Schmidt, L.S.; Linehan, W.M. Genetic predisposition to kidney cancer. Semin Oncol. 2016, 43, 566–574. [Google Scholar] [CrossRef]
- Chau, C.; van Doorn, R.; van Poppelen, N.M.; van der Stoep, N.; Mensenkamp, A.R.; Sijmons, R.H.; et al. Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: path to Identification and a Proposal for Genetic Screening Guidelines. Cancers (Basel). 2019, 11. [Google Scholar] [CrossRef]
- Carbone, M.; Pass, H.I.; Ak, G.; Alexander, H.R., Jr.; Baas, P.; Baumann, F.; et al. Medical and Surgical Care of Patients With Mesothelioma and Their Relatives Carrying Germline BAP1 Mutations. J Thorac Oncol. 2022, 17, 873–889. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.W.; An, J.Y.; Gomella, P.T.; Gautam, R.; Ricketts, C.J.; Vocke, C.D.; et al. Growth rates of genetically defined renal tumors: implications for active surveillance and intervention. J Clin Oncol. 2020, 38, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.H.; Lee, W.Y. Succinate dehydrogenase-deficient renal cell carcinoma. Arch Pathol Lab Med. 2019, 143, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J.; Pachter, N.S.; Chou, A.; Young, B.; Clarkson, A.; Tucker, K.M.; et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol. 2011, 35, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.K.; Luchtel, R.A.; Machha, V.; Tischer, A.; Zou, Y.; Pradhan, K.; et al. Functional succinate dehydrogenase deficiency is a common adverse feature of clear cell renal cancer. Proc Natl Acad Sci U S A. 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Milionis, V.; Goutas, D.; Vlachodimitropoulos, D.; Katsoulas, N.; Kyriazis, I.D.; Liatsikos, E.N.; et al. SDH-deficient renal cell carcinoma: a case report associated with a novel germline mutation. Clin Case Rep. 2021, 9, e04605. [Google Scholar] [CrossRef] [PubMed]
- Wilczek, Y.; Sachdeva, A.; Turner, H.; Veeratterapillay, R. SDH-deficient renal cell carcinoma: a clinicopathological analysis highlighting the role of genetic counselling. Ann R Coll Surg Engl. 2021, 103, e20-e2. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, N.; Yorita, K.; Nagasaki, M.; Harada, Y.; Ohe, C.; Jeruc, J.; et al. Review of succinate dehydrogenase-deficient renal cell carcinoma with focus on clinical and pathobiological aspects. Pol J Pathol. 2016, 67, 3–7. [Google Scholar] [CrossRef]
- van der Tuin, K.; Tops, C.M.J.; Adank, M.A.; Cobben, J.M.; Hamdy, N.A.T.; Jongmans, M.C.; et al. CDC73-Related Disorders: Clinical Manifestations and Case Detection in Primary Hyperparathyroidism. J Clin Endocrinol Metab. 2017, 102, 4534–4540. [Google Scholar] [CrossRef]
- Iacobone, M.; Carnaille, B.; Palazzo, F.F.; Vriens, M. Hereditary hyperparathyroidism--a consensus report of the European Society of Endocrine Surgeons (ESES). Langenbecks Arch Surg. 2015, 400, 867–886. [Google Scholar] [CrossRef] [PubMed]

This is an open access article under the terms of a license that permits non-commercial use, provided the original work is properly cited. © 2022 The Authors. Société Internationale d'Urologie Journal, published by the Société Internationale d'Urologie, Canada.
Share and Cite
Maranchie, J.K.; Shuch, B.M.; Bratslavsky, G.; Maher, E.R. 2022 WUOF/SIU International Consultation on Urological Diseases: Hereditary Renal Cell Carcinoma Syndromes. Soc. Int. Urol. J. 2022, 3, 397-406. https://doi.org/10.48083/CBTO5325
Maranchie JK, Shuch BM, Bratslavsky G, Maher ER. 2022 WUOF/SIU International Consultation on Urological Diseases: Hereditary Renal Cell Carcinoma Syndromes. Société Internationale d’Urologie Journal. 2022; 3(6):397-406. https://doi.org/10.48083/CBTO5325
Chicago/Turabian StyleMaranchie, Jodi K., Brian M. Shuch, Gennady Bratslavsky, and Eamonn R. Maher. 2022. "2022 WUOF/SIU International Consultation on Urological Diseases: Hereditary Renal Cell Carcinoma Syndromes" Société Internationale d’Urologie Journal 3, no. 6: 397-406. https://doi.org/10.48083/CBTO5325
APA StyleMaranchie, J. K., Shuch, B. M., Bratslavsky, G., & Maher, E. R. (2022). 2022 WUOF/SIU International Consultation on Urological Diseases: Hereditary Renal Cell Carcinoma Syndromes. Société Internationale d’Urologie Journal, 3(6), 397-406. https://doi.org/10.48083/CBTO5325