
Impacts of Electroconvulsive Therapy on the Neurometabolic Activity in a Mice Model of Depression: An Ex Vivo 1H-[13C]-NMR Spectroscopy Study

 ,
,
Abstract
:1. Introduction
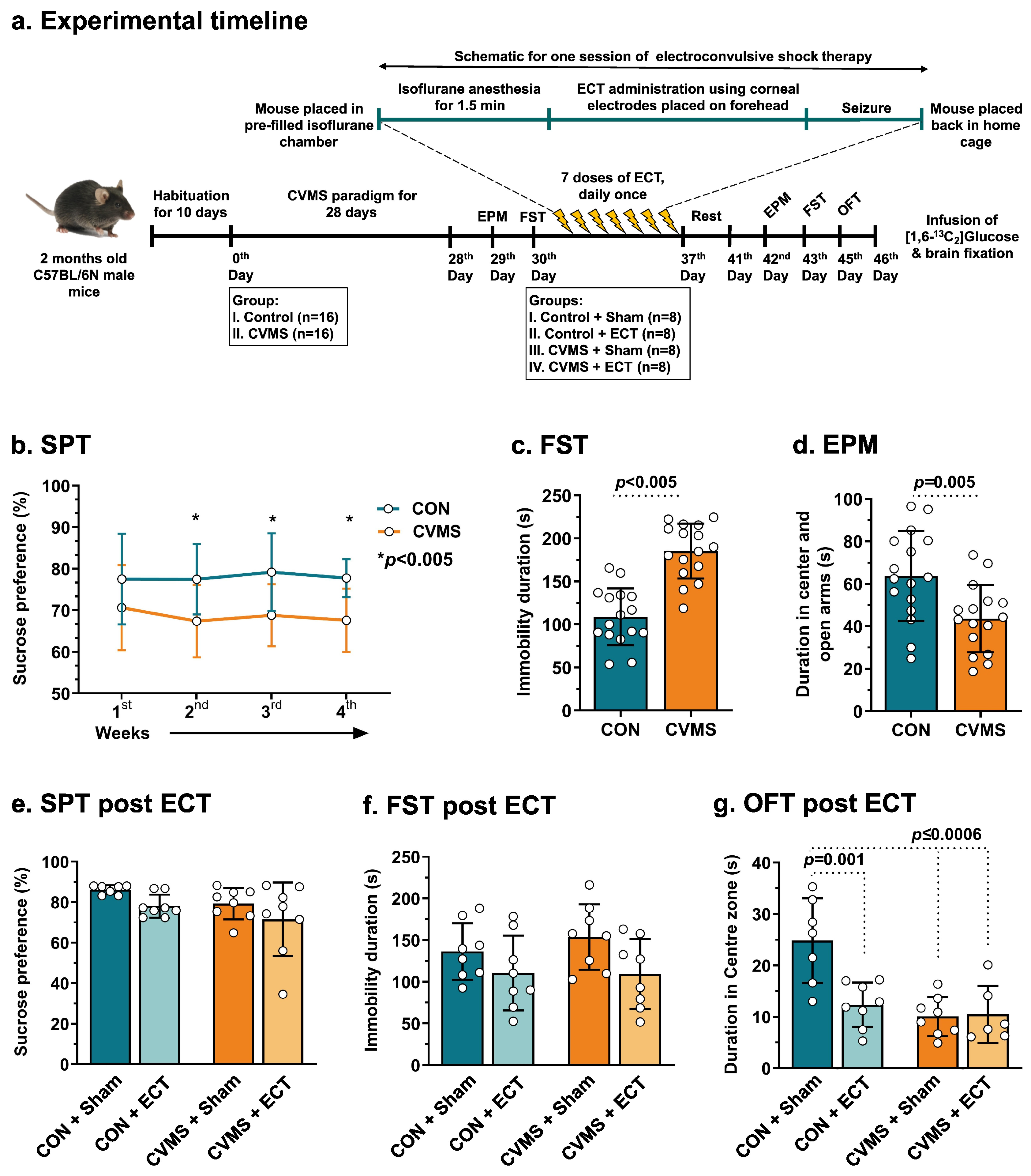
2. Materials and Methods
2.1. Animal Preparation
2.2. Chronic Variable Mild Stress (CVMS) Paradigm
2.3. Electroconvulsive Therapy (ECT) Procedure
2.4. Behavioral Analysis
2.4.1. Sucrose Preference Test (SPT)
2.4.2. Elevated-Plus Maze (EPM) Test
2.4.3. Open Field Test (OFT)
2.4.4. Forced-Swim Test (FST)
2.5. Infusion of [1,6-13C2]Glucose
2.6. Preparation of Brain Sample for NMR Analysis
2.7. NMR Analysis of Brain Samples
2.8. Determination of Rates of Glucose Oxidation
2.9. Statistical Analysis
3. Results and Discussion
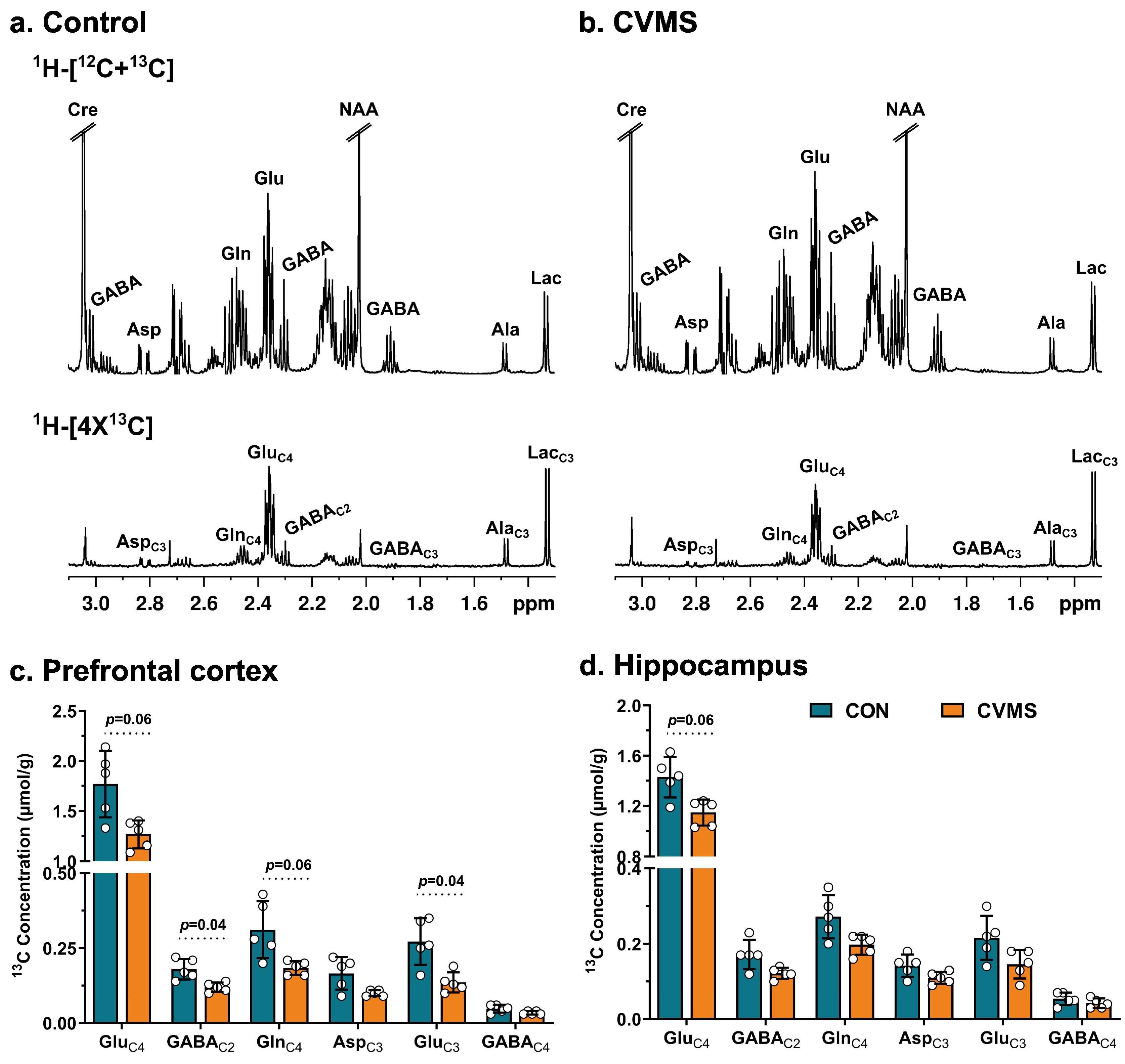
3.1. Impact of CVMS on Behavior and Metabolite Homeostasis
3.2. Labeling of Neurometabolites from [1,6-13C2]Glucose in CVMS Mice
3.3. Oxidative Glucose Metabolism in Glutamatergic and GABAergic Neurons in CVMS Mice
3.4. Effects of ECT on Depression- and Anxiety-like Phenotypes
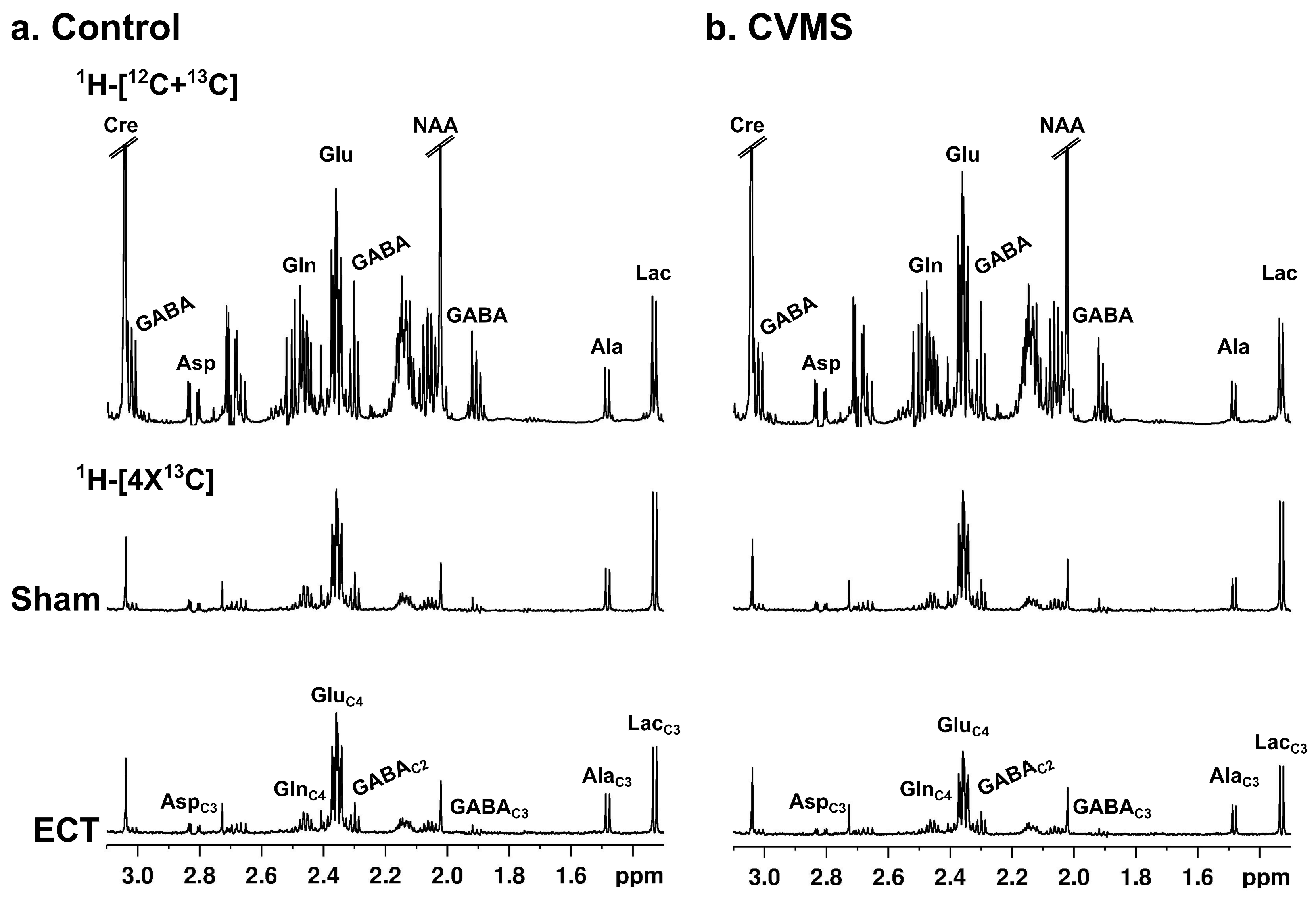
3.5. Impact of ECT on Neurometabolites Homeostasis
3.6. Effects of ECT on 13C-Labeling of Neurometabolites from [1,6-13C2]Glucose
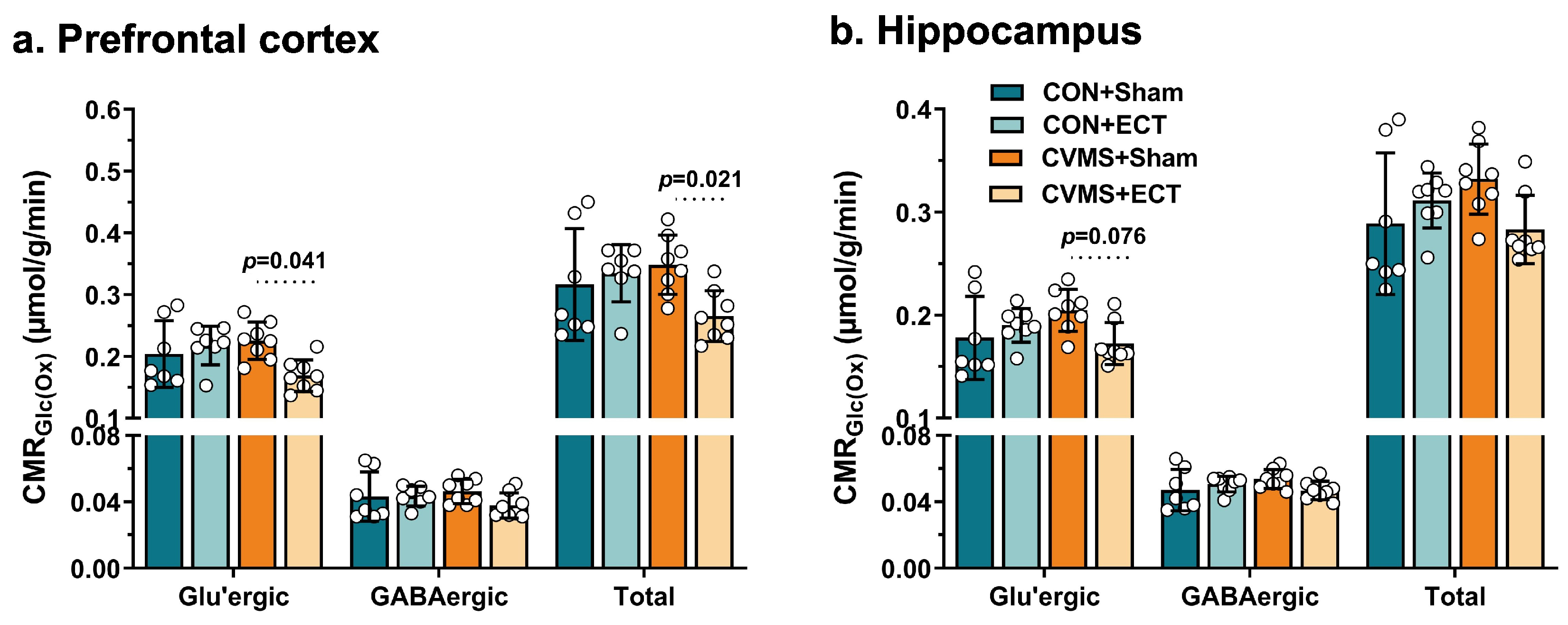
3.7. Effects of ECT on Metabolic Activity of Glutamatergic and GABAergic Neurons
4. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrman, H.; Patel, V.; Kieling, C.; Berk, M.; Buchweitz, C.; Cuijpers, P.; Furukawa, T.A.; Kessler, R.C.; Kohrt, B.A.; Maj, M.; et al. Time for United Action on Depression: A Lancet–World Psychiatric Association Commission. Lancet 2022, 399, 957–1022. [Google Scholar] [CrossRef] [PubMed]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major Depressive Disorder. Nat. Rev. Dis. Prim. 2016, 2, 16065. [Google Scholar] [CrossRef]
- Perez-Caballero, L.; Torres-Sanchez, S.; Romero-Lopez-Alberca, C.; Gonzalez-Saiz, F.; Mico, J.A.; Berrocoso, E. Monoaminergic System and Depression. Cell Tissue Res. 2019, 377, 107–113. [Google Scholar] [CrossRef]
- Voineskos, D.; Daskalakis, Z.J.; Blumberger, D.M. Management of Treatment-Resistant Depression: Challenges and Strategies. NDT 2020, 16, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, R.T.; Kellner, C.H. Electroconvulsive Therapy. N. Engl. J. Med. 2022, 386, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Cano, M.; Martínez-Zalacaín, I.; Bernabéu-Sanz, Á.; Contreras-Rodríguez, O.; Hernández-Ribas, R.; Via, E.; De Arriba-Arnau, A.; Gálvez, V.; Urretavizcaya, M.; Pujol, J.; et al. Brain Volumetric and Metabolic Correlates of Electroconvulsive Therapy for Treatment-Resistant Depression: A Longitudinal Neuroimaging Study. Transl. Psychiatry 2017, 7, e1023. [Google Scholar] [CrossRef] [PubMed]
- Erchinger, V.J.; Ersland, L.; Aukland, S.M.; Abbott, C.C.; Oltedal, L. Magnetic Resonance Spectroscopy in Depressed Subjects Treated with Electroconvulsive Therapy—A Systematic Review of Literature. Front. Psychiatry 2021, 12, 608857. [Google Scholar] [CrossRef]
- Maynard, K.R.; Hobbs, J.W.; Rajpurohit, S.K.; Martinowich, K. Electroconvulsive Seizures Influence Dendritic Spine Morphology and BDNF Expression in a Neuroendocrine Model of Depression. Brain Stimul. 2018, 11, 856–859. [Google Scholar] [CrossRef]
- Dukart, J.; Regen, F.; Kherif, F.; Colla, M.; Bajbouj, M.; Heuser, I.; Frackowiak, R.S.; Draganski, B. Electroconvulsive Therapy-Induced Brain Plasticity Determines Therapeutic Outcome in Mood Disorders. Proc. Natl. Acad. Sci. USA 2014, 111, 1156–1161. [Google Scholar] [CrossRef]
- Hosokawa, T.; Momose, T.; Kasai, K. Brain Glucose Metabolism Difference between Bipolar and Unipolar Mood Disorders in Depressed and Euthymic States. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 243–250. [Google Scholar] [CrossRef]
- Li, C.-T.; Su, T.-P.; Wang, S.-J.; Tu, P.-C.; Hsieh, J.-C. Prefrontal Glucose Metabolism in Medication-Resistant Major Depression. Br. J. Psychiatry 2015, 206, 316–323. [Google Scholar] [CrossRef]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated Energy Budgets for Neural Computation in the Neocortex and Cerebellum. J. Cereb. Blood Flow Metab. 2012, 32, 1222–1232. [Google Scholar] [CrossRef]
- Godfrey, K.E.M.; Gardner, A.C.; Kwon, S.; Chea, W.; Muthukumaraswamy, S.D. Differences in Excitatory and Inhibitory Neurotransmitter Levels between Depressed Patients and Healthy Controls: A Systematic Review and Meta-Analysis. J. Psychiatr. Res. 2018, 105, 33–44. [Google Scholar] [CrossRef]
- Sarawagi, A.; Soni, N.D.; Patel, A.B. Glutamate and GABA Homeostasis and Neurometabolism in Major Depressive Disorder. Front. Psychiatry 2021, 12, 637863. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Jiang, L.; De Feyter, H.M.; Fasula, M.; Krystal, J.H.; Rothman, D.L.; Mason, G.F.; Sanacora, G. Glutamate Metabolism in Major Depressive Disorder. AJP 2014, 171, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Adusumilli, M.; Deolal, P.; Mason, G.F.; Kumar, A.; Patel, A.B. Impaired Neuronal and Astroglial Metabolic Activity in Chronic Unpredictable Mild Stress Model of Depression: Reversal of Behavioral and Metabolic Deficit with Lanicemine. Neurochem. Int. 2020, 137, 104750. [Google Scholar] [CrossRef] [PubMed]
- Veeraiah, P.; Noronha, J.M.; Maitra, S.; Bagga, P.; Khandelwal, N.; Chakravarty, S.; Kumar, A.; Patel, A.B. Dysfunctional Glutamatergic and γ-Aminobutyric Acidergic Activities in Prefrontal Cortex of Mice in Social Defeat Model of Depression. Biol. Psychiatry 2014, 76, 231–238. [Google Scholar] [CrossRef]
- Brody, A.L.; Saxena, S.; Silverman, D.H.S.; Fairbanks, L.A.; Phelps, M.E.; Huang, S.-C.; Wu, H.-M.; Maidment, K.; Baxter, L.R.; Alborzian, S. Brain Metabolic Changes in Major Depressive Disorder from Pre- to Post-Treatment with Paroxetine. Psychiatry Res. Neuroimaging 1999, 91, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.H. Changes in Regional Brain Glucose Metabolism Measured with Positron Emission Tomography after Paroxetine Treatment of Major Depression. Am. J. Psychiatry 2001, 158, 899–905. [Google Scholar] [CrossRef]
- Mayberg, H.S.; Brannan, S.K.; Tekell, J.L.; Silva, J.A.; Mahurin, R.K.; McGinnis, S.; Jerabek, P.A. Regional Metabolic Effects of Fluoxetine in Major Depression: Serial Changes and Relationship to Clinical Response. Biol. Psychiatry 2000, 48, 830–843. [Google Scholar] [CrossRef]
- Mishra, P.K.; Kumar, A.; Behar, K.L.; Patel, A.B. Subanesthetic Ketamine Reverses Neuronal and Astroglial Metabolic Activity Deficits in a Social Defeat Model of Depression. J. Neurochem. 2018, 146, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, C.G.; De Feyter, H.M.; Averill, L.A.; Jiang, L.; Averill, C.L.; Chowdhury, G.M.I.; Purohit, P.; De Graaf, R.A.; Esterlis, I.; Juchem, C.; et al. The Effects of Ketamine on Prefrontal Glutamate Neurotransmission in Healthy and Depressed Subjects. Neuropsychopharmacol 2018, 43, 2154–2160. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.E.; Schmidt, M.E.; Matochik, J.A.; Stoddard, E.P.; Potter, W.Z. The Effects of ECT on Brain Glucose: A Pilot FDG PET Study. J. ECT 2001, 17, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Nobler, M.S.; Oquendo, M.A.; Kegeles, L.S.; Malone, K.M.; Campbell, C.; Sackeim, H.A.; Mann, J.J. Decreased Regional Brain Metabolism After ECT. AJP 2001, 158, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Gbyl, K.; Lindberg, U.; Wiberg Larsson, H.B.; Rostrup, E.; Videbech, P. Cerebral Perfusion Is Related to Antidepressant Effect and Cognitive Side Effects of Electroconvulsive Therapy. Brain Stimul. 2022, 15, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Duthie, A.C.; Perrin, J.S.; Bennett, D.M.; Currie, J.; Reid, I.C. Anticonvulsant Mechanisms of Electroconvulsive Therapy and Relation to Therapeutic Efficacy. J. ECT 2015, 31, 173–178. [Google Scholar] [CrossRef]
- Sackeim, H.A. The Anticonvulsant Hypothesis of the Mechanisms of Action of ECT: Current Status. J. ECT 1999, 15, 5–26. [Google Scholar] [CrossRef]
- McCormick, L.M.; Boles Ponto, L.L.; Pierson, R.K.; Johnson, H.J.; Magnotta, V.; Brumm, M.C. Metabolic Correlates of Antidepressant and Antipsychotic Response in Patients with Psychotic Depression Undergoing Electroconvulsive Therapy. J. ECT 2007, 23, 265–273. [Google Scholar] [CrossRef]
- Suwa, T.; Namiki, C.; Takaya, S.; Oshita, A.; Ishizu, K.; Fukuyama, H.; Suga, H.; Murai, T. Corticolimbic Balance Shift of Regional Glucose Metabolism in Depressed Patients Treated with ECT. J. Affect. Disord. 2012, 136, 1039–1046. [Google Scholar] [CrossRef]
- Yuuki, N.; Ida, I.; Oshima, A.; Kumano, H.; Takahashi, K.; Fukuda, M.; Oriuchi, N.; Endo, K.; Matsuda, H.; Mikuni, M. HPA Axis Normalization, Estimated by DEX/CRH Test, but Less Alteration on Cerebral Glucose Metabolism in Depressed Patients Receiving ECT after Medication Treatment Failures. Acta Psychiatr. Scand. 2005, 112, 257–265. [Google Scholar] [CrossRef]
- Fitzpatrick, S.M.; Hetherington, H.P.; Behar, K.L.; Shulman, R.G. The Flux from Glucose to Glutamate in the Rat Brain in Vivo as Determined by 1-Observed, 13C-Edited NMR Spectroscopy. J. Cereb. Blood Flow Metab. 1990, 10, 170–179. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, R.A.; Brown, P.B.; Mason, G.F.; Rothman, D.L.; Behar, K.L. Detection of [1,6-13C2]-Glucose Metabolism in Rat Brain by in Vivo 1H-[13C]-NMR Spectroscopy. Magn. Reson. Med. 2003, 49, 37–46. [Google Scholar] [CrossRef]
- De Graaf, R.A.; Mason, G.F.; Patel, A.B.; Behar, K.L.; Rothman, D.L. In vivo1H-[13C]-NMR Spectroscopy of Cerebral Metabolism. NMR Biomed. 2003, 16, 339–357. [Google Scholar] [CrossRef]
- Patel, A.B.; Tiwari, V.; Veeraiah, P.; Saba, K. Increased Astroglial Activity and Reduced Neuronal Function across Brain in AβPP-PS1 Mouse Model of Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2018, 38, 1213–1226. [Google Scholar] [CrossRef]
- Rothman, D.L.; Graaf, R.A.; Hyder, F.; Mason, G.F.; Behar, K.L.; De Feyter, H.M. In Vivo 13C and 1H-[13C] MRS Studies of Neuroenergetics and Neurotransmitter Cycling, Applications to Neurological and Psychiatric Disease and Brain Cancer. NMR Biomed. 2019, 32, e4172. [Google Scholar] [CrossRef] [PubMed]
- Willner, P.; Muscat, R.; Papp, M. Chronic Mild Stress-Induced Anhedonia: A Realistic Animal Model of Depression. Neurosci. Biobehav. Rev. 1992, 16, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Theilmann, W.; Löscher, W.; Socala, K.; Frieling, H.; Bleich, S.; Brandt, C. A New Method to Model Electroconvulsive Therapy in Rats with Increased Construct Validity and Enhanced Translational Value. J. Psychiatr. Res. 2014, 53, 94–98. [Google Scholar] [CrossRef]
- Liu, M.-Y.; Yin, C.-Y.; Zhu, L.-J.; Zhu, X.-H.; Xu, C.; Luo, C.-X.; Chen, H.; Zhu, D.-Y.; Zhou, Q.-G. Sucrose Preference Test for Measurement of Stress-Induced Anhedonia in Mice. Nat. Protoc. 2018, 13, 1686–1698. [Google Scholar] [CrossRef]
- Walf, A.A.; Frye, C.A. The Use of the Elevated plus Maze as an Assay of Anxiety-Related Behavior in Rodents. Nat. Protoc. 2007, 2, 322–328. [Google Scholar] [CrossRef]
- Gould, T.D.; Dao, D.T.; Kovacsics, C.E. The Open Field Test. In Mood and Anxiety Related Phenotypes in Mice; Gould, T.D., Ed.; Neuromethods; Humana Press: Totowa, NJ, USA, 2009; Volume 42, pp. 1–20. ISBN 978-1-60761-302-2. [Google Scholar]
- Juszczak, G.R.; Lisowski, P.; Śliwa, A.T.; Swiergiel, A.H. Computer Assisted Video Analysis of Swimming Performance in a Forced Swim Test: Simultaneous Assessment of Duration of Immobility and Swimming Style in Mice Selected for High and Low Swim-Stress Induced Analgesia. Physiol. Behav. 2008, 95, 400–407. [Google Scholar] [CrossRef]
- Tiwari, V.; Ambadipudi, S.; Patel, A.B. Glutamatergic and GABAergic TCA Cycle and Neurotransmitter Cycling Fluxes in Different Regions of Mouse Brain. J. Cereb. Blood Flow Metab. 2013, 33, 1523–1531. [Google Scholar] [CrossRef]
- Roy, D.; Puvvada, M.; Kapanaiah, S.K.T.; Patel, A.B. Enhanced Cortical Metabolic Activity in Females and Males of a Slow Progressing Mouse Model of Amyotrophic Lateral Sclerosis. Neurochem. Res. 2022, 47, 1765–1777. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, R.A.; Chowdhury, G.M.I.; Brown, P.B.; Rothman, D.L.; Behar, K.L. In Situ 3D Magnetic Resonance Metabolic Imaging of Microwave-Irradiated Rodent Brain: A New Tool for Metabolomics Research. J. Neurochem. 2009, 109, 494–501. [Google Scholar] [CrossRef]
- Patel, A.B.; Rothman, D.L.; Cline, G.W.; Behar, K.L. Glutamine Is the Major Precursor for GABA Synthesis in Rat Neocortex in Vivo Following Acute GABA-Transaminase Inhibition. Brain Res. 2001, 919, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Bagga, P.; Behar, K.L.; Mason, G.F.; De Feyter, H.M.; Rothman, D.L.; Patel, A.B. Characterization of Cerebral Glutamine Uptake from Blood in the Mouse Brain: Implications for Metabolic Modeling of 13 C NMR Data. J. Cereb. Blood Flow Metab. 2014, 34, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; De Graaf, R.A.; Mason, G.F.; Rothman, D.L.; Shulman, R.G.; Behar, K.L. The Contribution of GABA to Glutamate/Glutamine Cycling and Energy Metabolism in the Rat Cortex in Vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 5588–5593. [Google Scholar] [CrossRef] [PubMed]
- Unal, G.; Canbeyli, R. Psychomotor Retardation in Depression: A Critical Measure of the Forced Swim Test. Behav. Brain Res. 2019, 372, 112047. [Google Scholar] [CrossRef]
- Draganov, M.; Vives-Gilabert, Y.; De Diego-Adeliño, J.; Vicent-Gil, M.; Puigdemont, D.; Portella, M.J. Glutamatergic and GABA-Ergic Abnormalities in First-Episode Depression. A 1-Year Follow-up 1H-MR Spectroscopic Study. J. Affect. Disord. 2020, 266, 572–577. [Google Scholar] [CrossRef]
- Portella, M.J.; de Diego-Adelino, J.; Gomez-Anson, B.; Morgan-Ferrando, R.; Vives, Y.; Puigdemont, D.; Perez-Egea, R.; Ruscalleda, J.; Enric, A.; Perez, V. Ventromedial Prefrontal Spectroscopic Abnormalities over the Course of Depression: A Comparison among First Episode, Remitted Recurrent and Chronic Patients. J. Psychiatr. Res. 2011, 45, 427–434. [Google Scholar] [CrossRef]
- Hasler, G.; Meyers, N.; Shen, J.; Drevets, W.C. Reduced Prefrontal Glutamate/Glutamine and γ-Aminobutyric Acid Levels in Major Depression Determined Using Proton Magnetic Resonance Spectroscopy. Arch. Gen. Psychiatry 2007, 64, 193–200. [Google Scholar] [CrossRef]
- Jollant, F.; Richard-Devantoy, S.; Ding, Y.; Turecki, G.; Bechara, A.; Near, J. Prefrontal Inositol Levels and Implicit Decision-Making in Healthy Individuals and Depressed Patients. Eur. Neuropsychopharmacol. 2016, 26, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, M.K.; Near, J.; Blicher, A.B.; Videbech, P.; Blicher, J.U. Magnetic Resonance (MR) Spectroscopic Measurement of γ-Aminobutyric Acid (GABA) in Major Depression before and after Electroconvulsive Therapy. Acta Neuropsychiatr. 2019, 31, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Moussawi, K.; Riegel, A.; Nair, S.; Kalivas, P.W. Extracellular Glutamate: Functional Compartments Operate in Different Concentration Ranges. Front. Syst. Neurosci. 2011, 5, 94. [Google Scholar] [CrossRef] [PubMed]
- Chiappelli, J.; Adhikari, B.M.; Kvarta, M.D.; Bruce, H.A.; Goldwaser, E.L.; Ma, Y.; Chen, S.; Ament, S.; Shuldiner, A.R.; Mitchell, B.D.; et al. Depression, Stress and Regional Cerebral Blood Flow. J. Cereb. Blood Flow Metab. 2023, 43, 791–800. [Google Scholar] [CrossRef]
- Monkul, E.S.; Silva, L.A.P.; Narayana, S.; Peluso, M.A.M.; Zamarripa, F.; Nery, F.G.; Najt, P.; Li, J.; Lancaster, J.L.; Fox, P.T.; et al. Abnormal Resting State Corticolimbic Blood Flow in Depressed Unmedicated Patients with Major Depression: A 15O-H2O PET Study. Hum. Brain Mapp. 2012, 33, 272–279. [Google Scholar] [CrossRef]
- Hyder, F.; Patel, A.B.; Gjedde, A.; Rothman, D.L.; Behar, K.L.; Shulman, R.G. Neuronal–Glial Glucose Oxidation and Glutamatergic–GABAergic Function. J. Cereb. Blood Flow Metab. 2006, 26, 865–877. [Google Scholar] [CrossRef]
- Rothman, D.L.; Behar, K.L.; Dienel, G.A. Mechanistic Stoichiometric Relationship between the Rates of Neurotransmission and Neuronal Glucose Oxidation: Reevaluation of and Alternatives to the Pseudo-malate-aspartate Shuttle Model. J. Neurochem. 2024, 168, 555–591. [Google Scholar] [CrossRef]
- Zhu, X.; Ye, G.; Wang, Z.; Luo, J.; Hao, X. Sub-Anesthetic Doses of Ketamine Exert Antidepressant-like Effects and Upregulate the Expression of Glutamate Transporters in the Hippocampus of Rats. Neurosci. Lett. 2017, 639, 132–137. [Google Scholar] [CrossRef]
- Antila, H.; Ryazantseva, M.; Popova, D.; Sipilä, P.; Guirado, R.; Kohtala, S.; Yalcin, I.; Lindholm, J.; Vesa, L.; Sato, V.; et al. Isoflurane Produces Antidepressant Effects and Induces TrkB Signaling in Rodents. Sci. Rep. 2017, 7, 7811. [Google Scholar] [CrossRef]
- Zhang, S.-S.; Tian, Y.-H.; Jin, S.-J.; Wang, W.-C.; Zhao, J.-X.; Si, X.-M.; Zhang, L.; Xu, H.; Jin, J.-Y. Isoflurane Produces Antidepressant Effects Inducing BDNF-TrkB Signaling in CUMS Mice. Psychopharmacology 2019, 236, 3301–3315. [Google Scholar] [CrossRef]
- Weeks, H.R.; Tadler, S.C.; Smith, K.W.; Iacob, E.; Saccoman, M.; White, A.T.; Landvatter, J.D.; Chelune, G.J.; Suchy, Y.; Clark, E.; et al. Antidepressant and Neurocognitive Effects of Isoflurane Anesthesia versus Electroconvulsive Therapy in Refractory Depression. PLoS ONE 2013, 8, e69809. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.L.; Zanos, P.; Wang, L.; Elmer, G.I.; Gould, T.D.; Shepard, P.D. Isoflurane but Not Halothane Prevents and Reverses Helpless Behavior: A Role for EEG Burst Suppression? Int. J. Neuropsychopharmacol. 2018, 21, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Michael, N.; Erfurth, A.; Ohrmann, P.; Arolt, V.; Heindel, W.; Pfleiderer, B. Neurotrophic Effects of Electroconvulsive Therapy: A Proton Magnetic Resonance Study of the Left Amygdalar Region in Patients with Treatment-Resistant Depression. Neuropsychopharmacology 2003, 28, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Michael, N.; Erfurth, A.; Ohrmann, P.; Arolt, V.; Heindel, W.; Pfleiderer, B. Metabolic Changes within the Left Dorsolateral Prefrontal Cortex Occurring with Electroconvulsive Therapy in Patients with Treatment Resistant Unipolar Depression. Psychol. Med. 2003, 33, 1277–1284. [Google Scholar] [CrossRef]
- Pfleiderer, B.; Michael, N.; Erfurth, A.; Ohrmann, P.; Hohmann, U.; Wolgast, M.; Fiebich, M.; Arolt, V.; Heindel, W. Effective Electroconvulsive Therapy Reverses Glutamate/Glutamine Deficit in the Left Anterior Cingulum of Unipolar Depressed Patients. Psychiatry Res. Neuroimaging 2003, 122, 185–192. [Google Scholar] [CrossRef]
- Sanacora, G.; Mason, G.F.; Rothman, D.L.; Hyder, F.; Ciarcia, J.J.; Ostroff, R.B.; Berman, R.M.; Krystal, J.H. Increased Cortical GABA Concentrations in Depressed Patients Receiving ECT. AJP 2003, 160, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Njau, S.; Joshi, S.H.; Leaver, A.M.; Vasavada, M.; Van Fleet, J.; Espinoza, R.; Narr, K.L. Variations in Myo-Inositol in Fronto-Limbic Regions and Clinical Response to Electroconvulsive Therapy in Major Depression. J. Psychiatr. Res. 2016, 80, 45–51. [Google Scholar] [CrossRef]
- Rimmerman, N.; Verdiger, H.; Goldenberg, H.; Naggan, L.; Robinson, E.; Kozela, E.; Gelb, S.; Reshef, R.; Ryan, K.M.; Ayoun, L.; et al. Microglia and Their LAG3 Checkpoint Underlie the Antidepressant and Neurogenesis-Enhancing Effects of Electroconvulsive Stimulation. Mol. Psychiatry 2022, 27, 1120–1135. [Google Scholar] [CrossRef]
- Zhang, J.; Narr, K.L.; Woods, R.P.; Phillips, O.R.; Alger, J.R.; Espinoza, R.T. Glutamate Normalization with ECT Treatment Response in Major Depression. Mol. Psychiatry 2013, 18, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Ende, G.; Braus, D.F.; Walter, S.; Weber-Fahr, W.; Henn, F.A. The Hippocampus in Patients Treated With Electroconvulsive Therapy: A Proton Magnetic Resonance Spectroscopic Imaging Study. Arch. Gen. Psychiatry 2000, 57, 937. [Google Scholar] [CrossRef]
- Leaver, A.M.; Vasavada, M.; Joshi, S.H.; Wade, B.; Woods, R.P.; Espinoza, R.; Narr, K.L. Mechanisms of Antidepressant Response to Electroconvulsive Therapy Studied with Perfusion Magnetic Resonance Imaging. Biol. Psychiatry 2019, 85, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, V.; Hadzi-Pavlovic, D.; Waite, S.; Loo, C.K. Seizure Threshold Increases Can Be Predicted by EEG Quality in Right Unilateral Ultrabrief ECT. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 267, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Ma, V.; Gonzalez, C.; Chapman, A.; Printz, D.; Voytek, B.; Soltani, M. Clinical EEG Slowing Induced by Electroconvulsive Therapy Is Better Described by Increased Frontal Aperiodic Activity. Transl. Psychiatry 2023, 13, 348. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.Z.; Reininghaus, B.; Enzinger, C.; Ebner, C.; Hofmann, P.; Kapfhammer, H.P. Changes in Brain Metabolism after ECT–Positron Emission Tomography in the Assessment of Changes in Glucose Metabolism Subsequent to Electroconvulsive Therapy—Lessons, Limitations and Future Applications. J. Affect. Disord. 2008, 106, 203–208. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glu | GABA | Gln | Asp | NAA | Lac | m-Ino | Tau | GPC | Cre | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PFC | CON | Sham (n = 7) | 13.2 ± 0.8 | 3.6 ± 0.3 | 6.3 ± 0.3 | 3.0 ± 0.2 | 7.4 ± 0.4 | 1.8 ± 0.5 | 8.4 ± 0.4 | 12.9 ± 0.7 | 1.4 ± 0.1 | 14.0 ± 0.7 |
| ECT (n = 8) | 13.5 ± 1.2 | 3.6 ± 0.2 | 6.2 ± 0.6 | 2.9 ± 0.3 | 7.4 ± 0.6 | 1.9 ± 0.4 | 8.7 ± 0.5 | 12.9 ± 0.9 | 1.4 ± 0.0 | 14.2 ± 1.0 | ||
| CVMS | Sham (n = 8) | 12.5 ± 1.1 | 3.5 ± 0.1 | 5.8 ± 0.4 | 2.9 ± 0.2 | 7.1 ± 0.5 | 1.8 ± 0.6 | 8.2 ± 0.3 | 12.9 ± 0.4 | 1.4 ± 0.1 | 13.5 ± 0.6 | |
| ECT (n = 8) | 11.7 ± 0.9 ## | 3.6 ± 0.2 | 5.8 ± 0.3 | 2.7 ± 0.2 | 6.8 ± 0.4 | 1.3 ± 0.1 | 8.4 ± 0.6 | 12.9 ± 0.6 | 1.4 ± 0.1 | 13.3 ± 0.6 | ||
| HIP | CON | Sham (n = 7) | 12.2 ± 0.7 | 3.6 ± 0.2 | 5.7 ± 0.5 | 2.8 ± 0.2 | 6.7 ± 0.5 | 1.6 ± 0.2 | 9.8 ± 0.7 | 10.0 ± 0.6 | 1.4 ± 0.1 | 15.3 ± 0.6 |
| ECT (n = 8) | 12.5 ± 0.5 | 3.4 ± 0.1 | 5.7 ± 0.4 | 2.6 ± 0.1 | 6.6 ± 0.2 | 1.6 ± 0.2 | 10.0 ± 0.4 | 9.9 ± 0.4 | 1.4 ± 0.1 | 15.2 ± 0.6 | ||
| CVMS | Sham (n = 8) | 12.1 ± 0.5 | 3.2 ± 0.1 * | 5.5 ± 0.4 | 2.8 ± 0.2 | 6.5 ± 0.3 | 1.5 ± 0.1 | 9.1 ± 0.4 * | 10.1 ± 0.2 | 1.4 ± 0.1 | 14.9 ± 0.4 | |
| ECT (n = 8) | 11.8 ± 0.6 | 3.4 ± 0.3 | 5.6 ± 0.2 | 2.7 ± 0.2 | 6.8 ± 0.3 | 1.3 ± 0.2 # | 9.6 ± 0.5 | 10.0 ± 0.4 | 1.4 ± 0.1 | 15.2 ± 0.8 |
| GluC4 | GABAC2 | GlnC4 | AspC3 | GluC3 | GABAC4 | AlaC3 | LacC3 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| PFC | CON | Sham (n = 7) | 1.88 ± 0.38 | 0.17 ± 0.06 | 0.27 ± 0.11 | 0.16 ± 0.06 | 0.25 ± 0.12 | 0.04 ± 0.02 | 0.16 ± 0.05 | 0.38 ± 0.20 |
| ECT (n = 8) | 2.08 ± 0.40 | 0.20 ± 0.04 | 0.29 ± 0.04 | 0.18 ± 0.05 | 0.28 ± 0.06 | 0.03 ± 0.01 | 0.16 ± 0.02 | 0.44 ± 0.14 | ||
| CVMS | Sham (n = 8) | 2.06 ± 0.27 | 0.18 ± 0.03 | 0.30 ± 0.05 | 0.17 ± 0.03 | 0.28 ± 0.06 | 0.05 ± 0.01 | 0.13 ± 0.02 | 0.40 ± 0.12 | |
| ECT (n = 8) | 1.59 ± 0.20 #$ | 0.15 ± 0.01 | 0.24 ± 0.04 | 0.13 ± 0.03 | 0.20 ± 0.05 | 0.04 ± 0.01 | 0.11 ± 0.05 | 0.23 ± 0.11 # | ||
| HIP | CON | Sham (n = 7) | 1.58 ± 0.30 | 0.21 ± 0.06 | 0.27 ± 0.08 | 0.16 ± 0.05 | 0.23 ± 0.08 | 0.05 ± 0.01 | 0.13 ± 0.01 | 0.30 ± 0.08 |
| ECT (n = 8) | 1.72 ± 0.20 | 0.23 ± 0.03 | 0.31 ± 0.05 | 0.18 ± 0.03 | 0.25 ± 0.03 | 0.05 ± 0.01 | 0.15 ± 0.01 | 0.39 ± 0.09 | ||
| CVMS | Sham (n = 8) | 1.77 ± 0.16 | 0.24 ± 0.03 | 0.31 ± 0.05 | 0.18 ± 0.02 | 0.28 ± 0.05 | 0.05 ± 0.01 | 0.13 ± 0.01 | 0.34 ± 0.05 | |
| ECT (n = 8) | 1.53 ± 0.20 | 0.20 ± 0.01 | 0.28 ± 0.04 | 0.16 ± 0.04 | 0.23 ± 0.02 | 0.06 ± 0.04 | 0.13 ± 0.02 | 0.26 ± 0.05 ## |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarawagi, A.; Wadnerkar, P.; Keluskar, V.; Ram, N.S.; Kumar, J.M.; Patel, A.B. Impacts of Electroconvulsive Therapy on the Neurometabolic Activity in a Mice Model of Depression: An Ex Vivo 1H-[13C]-NMR Spectroscopy Study. Neuroglia 2024, 5, 306-322. https://doi.org/10.3390/neuroglia5030021
Sarawagi A, Wadnerkar P, Keluskar V, Ram NS, Kumar JM, Patel AB. Impacts of Electroconvulsive Therapy on the Neurometabolic Activity in a Mice Model of Depression: An Ex Vivo 1H-[13C]-NMR Spectroscopy Study. Neuroglia. 2024; 5(3):306-322. https://doi.org/10.3390/neuroglia5030021
Chicago/Turabian StyleSarawagi, Ajay, Pratishtha Wadnerkar, Vrundika Keluskar, Narra Sai Ram, Jerald Mahesh Kumar, and Anant Bahadur Patel. 2024. "Impacts of Electroconvulsive Therapy on the Neurometabolic Activity in a Mice Model of Depression: An Ex Vivo 1H-[13C]-NMR Spectroscopy Study" Neuroglia 5, no. 3: 306-322. https://doi.org/10.3390/neuroglia5030021
APA StyleSarawagi, A., Wadnerkar, P., Keluskar, V., Ram, N. S., Kumar, J. M., & Patel, A. B. (2024). Impacts of Electroconvulsive Therapy on the Neurometabolic Activity in a Mice Model of Depression: An Ex Vivo 1H-[13C]-NMR Spectroscopy Study. Neuroglia, 5(3), 306-322. https://doi.org/10.3390/neuroglia5030021