
Harnessing Mitophagy for Therapeutic Advances in Aging and Chronic Neurodegenerative Diseases

Abstract
:1. Introduction
2. Mitophagy: Mechanism and Neuroprotection
3. Mitophagy and Aging
4. Mitophagy and Neuroinflammation
5. Therapeutic Interventions to Modulate Mitophagy
6. Challenges and Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DAMPs | Damage-associated proteins |
| CoQ10 | Coenzyme Q10 |
| PINK1/Parkin | PTEN-induced kinase 1 |
| BNIP3 BCL2 | Interacting protein 3 |
| NIX FUNDC1 | FUN14 domain containing 1LC3 |
| MDD | Major depressive disorder |
| Drp1 | Dynamin-related protein 1 |
| ROS | Reactive oxygen species |
| AMPK | AMP-activated protein kinase |
| mTOR | Mammalian/mechanistic target of rapamycin |
| OPA1 | Optic atrophy 1 |
| mtDNA | Mitochondrial DNA |
| FGF | Fibroblast growth factor |
| GDF | Growth differentiation factor |
| cGAS | Cyclic GMP-AMP synthase |
| -STING | Stimulator of interferon genes |
| NLRP3 | Nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing-3 |
| TLRs | Toll-like receptors |
| NLRs | NOD-like receptors |
| CNS | Central Nervous System |
| HMGB1 | High Mobility Group Box-1 |
| AD | Alzheimer’s Disease |
| PD | Parkinson’s Disease |
| HD | Huntington’s Disease |
| MS | Multiple Sclerosis |
| ALS | Amyotrophic lateral sclerosis |
| SIRT | Sirtuins |
| UA | Urolithin A |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CR | Caloric restriction |
| BDNF | Brain-derived neurotrophic factor |
| VEGF | Vascular endothelial growth factor |
References
- Rappe, A.; McWilliams, T.G. Mitophagy in the aging nervous system. Front. Cell Dev. Biol. 2022, 10, 978142. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.-S.Z.; Sun, Y.; Pearce, A.C.; Eleuteri, S.; Kemp, M.; Luckhurst, C.A.; Williams, R.; Mills, R.; Almond, S.; Burzynski, L.; et al. Knockout or inhibition of USP30 protects dopaminergic neurons in a Parkinson’s disease mouse model. Nat. Commun. 2023, 14, 7295. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Scheibye-Knudsen, M. MitophAging: Mitophagy in Aging and Disease. Front. Cell Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cao, H.; Zuo, C.; Gu, Z.; Huang, Y.; Miao, J.; Fu, Y.; Guo, Y.; Jiang, Y.; Wang, F. Mitochondrial dysfunction: A fatal blow in depression. Biomed. Pharmacother. 2023, 167, 115652. [Google Scholar] [CrossRef]
- Dong, W.-T.; Long, L.-H.; Deng, Q.; Liu, D.; Wang, J.-L.; Wang, F.; Chen, J.-G. Mitochondrial fission drives neuronal metabolic burden to promote stress susceptibility in male mice. Nat. Metab. 2023, 5, 2220–2236. [Google Scholar] [CrossRef]
- Charmpilas, N.; Fang, E.F.; Palikaras, K. Mitophagy and Neuroinflammation: A Compelling Interplay. Curr. Neuropharmacol. 2023, 21, 1477–1481. [Google Scholar] [CrossRef]
- Lautrup, S.; Lou, G.; Aman, Y.; Nilsen, H.; Tao, J.; Fang, E.F. Microglial mitophagy mitigates neuroinflammation in Alzheimer’s disease. Neurochem. Int. 2019, 129, 104469. [Google Scholar] [CrossRef]
- Qiu, J.; Chen, Y.; Zhuo, J.; Zhang, L.; Liu, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A promotes mitophagy and suppresses NLRP3 inflammasome activation in lipopolysaccharide-induced BV2 microglial cells and MPTP-induced Parkinson’s disease model. Neuropharmacology 2022, 207, 108963. [Google Scholar] [CrossRef]
- Chen, Q.; Fan, K.; Song, G.; Wang, X.; Zhang, J.; Chen, H.; Qin, X.; Lu, Y.; Qi, W. Rapamycin regulates osteogenic differentiation through Parkin-mediated mitophagy in rheumatoid arthritis. Int. Immunopharmacol. 2022, 113, 109407. [Google Scholar] [CrossRef]
- Zhang, Y.; Bai, J.; Cui, Z.; Li, Y.; Gao, Q.; Miao, Y.; Xiong, B. Polyamine metabolite spermidine rejuvenates oocyte quality by enhancing mitophagy during female reproductive aging. Nat. Aging 2023, 3, 1372–1386. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Behera, B.P.; Mishra, S.R.; Bhutia, S.K. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin. Cancer Biol. 2020, 66, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.-W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Z.; Zhang, S.; Zhang, T.; Liu, Y.; Zhang, L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics 2023, 13, 736–766. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Li, Z.; Zhao, M.; Wang, D.; Sun, Z.; Wen, P.; Dai, Y.; Gou, F.; Ji, Y.; et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases. Ageing Res. Rev. 2023, 84, 101817. [Google Scholar] [CrossRef]
- Borsche, M.; König, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Brüggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M.; et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 2020, 143, 3041–3051. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 20009. [Google Scholar] [CrossRef]
- Cossu, D.; Yokoyama, K.; Sechi, L.A.; Hattori, N. Potential of PINK1 and PARKIN Proteins as Biomarkers for Active Multiple Sclerosis: A Japanese Cohort Study. Front. Immunol. 2021, 12, 681386. [Google Scholar] [CrossRef]
- Ito, A.; Hashimoto, M.; Tanihata, J.; Matsubayashi, S.; Sasaki, R.; Fujimoto, S.; Kawamoto, H.; Hosaka, Y.; Ichikawa, A.; Kadota, T.; et al. Involvement of Parkin-mediated mitophagy in the pathogenesis of chronic obstructive pulmonary disease-related sarcopenia. J. Cachexia Sarcopenia Muscle 2022, 13, 1864–1882. [Google Scholar] [CrossRef]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zheng, J.; Wan, H.; Sun, Y.; Fu, S.; Liu, S.; He, B.; Cai, G.; Cao, Y.; Huang, H.; et al. A mitochondrial SCF-FBXL4 ubiquitin E3 ligase complex degrades BNIP3 and NIX to restrain mitophagy and prevent mitochondrial disease. EMBO J. 2023, 42, e113033. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Wang, J.; Zhang, D.; Wu, H.; Li, W.; Wei, H.; Ta, N.; Fan, Y.; Liu, Y.; et al. Mitophagy receptor FUNDC1 is regulated by PGC-1α/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 2021, 22, e50629. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef]
- Cho, K.S.; Lee, J.H.; Cho, J.; Cha, G.-H.; Song, G.J. Autophagy Modulators and Neuroinflammation. Curr. Med. Chem. 2020, 27, 955–982. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, J.; Li, D.; Ran, S.; Huang, J.; Chen, G. Morin exhibits a neuroprotective effect in MPTP-induced Parkinson’s disease model via TFEB/AMPK-mediated mitophagy. Phytomedicine 2023, 116, 154866. [Google Scholar] [CrossRef]
- Gravandi, M.M.; Fakhri, S.; Zarneshan, S.N.; Yarmohammadi, A.; Khan, H. Flavonoids modulate AMPK/PGC-1α and interconnected pathways toward potential neuroprotective activities. Metab. Brain Dis. 2021, 36, 1501–1521. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, Å.B. The role of mitochondrial fission in cardiovascular health and disease. Nat. Rev. Cardiol. 2022, 19, 723–736. [Google Scholar] [CrossRef]
- Jin, J.-Y.; Wei, X.-X.; Zhi, X.-L.; Wang, X.-H.; Meng, D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol. Sin. 2021, 42, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Singh, G.; Mishra, P.; Singh, A.; Kumar, A.; Sinha, N. Alteration in mitochondrial dynamics promotes the proinflammatory response of microglia and is involved in cerebellar dysfunction of young and aged mice following LPS exposure. Neurosci. Lett. 2023, 807, 137262. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhu, J.; Gao, Q.; Rebecchi, M.J.; Wang, Q.; Liu, L. Restoring Pharmacologic Preconditioning in the Aging Heart: Role of Mitophagy/Autophagy. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.-P.; Hussain, S.N.A.; Barreiro, E.; Gouspillou, G. Mitochondrial Dynamics and Mitophagy in Skeletal Muscle Health and Aging. Int. J. Mol. Sci. 2021, 22, 8179. [Google Scholar] [CrossRef] [PubMed]
- Mengel-From, J.; Thinggaard, M.; Dalgård, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum. Genet. 2014, 133, 1149–1159. [Google Scholar] [CrossRef]
- Oliveira, A.N.; Karmanova, L.; Murugavel, S.; Yanagawa, B.; Hood, D.A. Enhanced Mitochondrial Turnover in Aged Human Right Atrial Tissue. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Conte, M.; Ostan, R.; Fabbri, C.; Santoro, A.; Guidarelli, G.; Vitale, G.; Mari, D.; Sevini, F.; Capri, M.; Sandri, M.; et al. Human Aging and Longevity Are Characterized by High Levels of Mitokines. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 600–607. [Google Scholar] [CrossRef]
- Lin, M.; Liu, N.; Qin, Z.; Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef]
- Dagvadorj, J.; Mikulska-Ruminska, K.; Tumurkhuu, G.; Ratsimandresy, R.A.; Carriere, J.; Andres, A.M.; Marek-Iannucci, S.; Song, Y.; Chen, S.; Lane, M.; et al. Recruitment of pro-IL-1α to mitochondrial cardiolipin, via shared LC3 binding domain, inhibits mitophagy and drives maximal NLRP3 activation. Proc. Natl. Acad. Sci. USA 2021, 118, e2015632118. [Google Scholar] [CrossRef]
- Liu, J.; Wang, T.; He, K.; Xu, M.; Gong, J.-P. Cardiolipin inhibitor ameliorates the non-alcoholic steatohepatitis through suppressing NLRP3 inflammasome activation. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8158–8167. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front. Immunol. 2016, 7, 279. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. Small molecules that enhance mitophagy to delay aging and neurodegeneration. J. Cardiovasc. Aging 2022, 2, 45. [Google Scholar] [CrossRef] [PubMed]
- Coen, P.M.; Huo, Z.; Tranah, G.J.; Barnes, H.N.; Zhang, X.; Wolff, C.A.; Wu, K.; Cawthon, P.M.; Hepple, R.T.; Toledo, F.G.S.; et al. Autophagy gene expression in skeletal muscle of older individuals is associated with physical performance, muscle volume and mitochondrial function in the study of muscle, mobility and aging (SOMMA). Aging Cell 2024, 23, e14118. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.; Bhansali, A.; Walia, R.; Saikia, U.N.; Dhawan, V. Alterations in Mitochondrial Oxidative Stress and Mitophagy in Subjects with Prediabetes and Type 2 Diabetes Mellitus. Front. Endocrinol. 2017, 8, 347. [Google Scholar] [CrossRef] [PubMed]
- Veluthakal, R.; Esparza, D.; Hoolachan, J.M.; Balakrishnan, R.; Ahn, M.; Oh, E.; Jayasena, C.S.; Thurmond, D.C. Mitochondrial Dysfunction, Oxidative Stress, and Inter-Organ Miscommunications in T2D Progression. Int. J. Mol. Sci. 2024, 25, 1504. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Kitamura, Y.; Nomura, Y.; Segawa, T. Possible involvement of inhibitory GTP binding regulatory protein in alpha 2-adrenoceptor-mediated inhibition of adenylate cyclase activity in cerebral cortical membranes of rats. J. Neurochem. 1985, 45, 1504–1508. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Chang, P.; Li, H.; Hu, H.; Li, Y.; Wang, T. The Role of HDAC6 in Autophagy and NLRP3 Inflammasome. Front. Immunol. 2021, 12, 763831. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and Mitophagy Connection in Health and Disease. Int. J. Mol. Sci. 2020, 21, 4714. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhou, X.; Fang, L.; Dong, J.; Cui, L.; Li, J.; Meng, X.; Zhu, G.; Li, J.; Wang, H. PINK1/parkin-mediated mitophagy alleviates Staphylococcus aureus-induced NLRP3 inflammasome and NF-κB pathway activation in bovine mammary epithelial cells. Int. Immunopharmacol. 2022, 112, 109200. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.; Yu, L.; Qiao, G.; Qin, D.; Yuen-Kwan Law, B.; Ren, F.; Wu, J.; Wu, A. Mitophagy and cGAS–STING crosstalk in neuroinflammation. Acta Pharm. Sin. B 2024, 14, 3327–3361. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H.; Bohr, V.A. Signaling by cGAS-STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci. 2021, 44, 83–96. [Google Scholar] [CrossRef]
- Jiménez-Loygorri, J.I.; Villarejo-Zori, B.; Viedma-Poyatos, Á.; Zapata-Muñoz, J.; Benítez-Fernández, R.; Frutos-Lisón, M.D.; Tomás-Barberán, F.A.; Espín, J.C.; Area-Gómez, E.; Gomez-Duran, A.; et al. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat. Commun. 2024, 15, 830. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD + supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS–STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- Jiménez-Loygorri, J.I.; Boya, P. Aging STINGs: Mitophagy at the crossroads of neuroinflammation. Autophagy 2024, 20, 1684–1686. [Google Scholar] [CrossRef]
- Ghosh, D.; Singh, A.; Kumar, A.; Sinha, N. High mobility group box 1 (HMGB1) inhibition attenuates lipopolysaccharide-induced cognitive dysfunction and sickness-like behavior in mice. Immunol. Res. 2022, 70, 633–643. [Google Scholar] [CrossRef]
- Razali, K.; Mohd Nasir, M.H.; Kumar, J.; Mohamed, W.M.Y. Mitophagy: A Bridge Linking HMGB1 and Parkinson’s Disease Using Adult Zebrafish as a Model Organism. Brain Sci. 2023, 13, 1076. [Google Scholar] [CrossRef]
- Qu, L.; Chen, C.; Chen, Y.; Li, Y.; Tang, F.; Huang, H.; He, W.; Zhang, R.; Shen, L. High-Mobility Group Box 1 (HMGB1) and Autophagy in Acute Lung Injury (ALI): A Review. Med. Sci. Monit. 2019, 25, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Castellazzi, M.; Bonora, M.; Marchi, S.; Casetta, I.; Pugliatti, M.; Giorgi, C.; Granieri, E.; Pinton, P. Autophagy and mitophagy elements are increased in body fluids of multiple sclerosis-affected individuals. J. Neurol. Neurosurg. Psychiatry 2018, 89, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Chu, X.; Park, J.-H.; Zhu, Q.; Hussain, M.; Li, Z.; Madsen, H.B.; Yang, B.; Wei, Y.; Wang, Y.; et al. Urolithin A improves Alzheimer’s disease cognition and restores mitophagy and lysosomal functions. Alzheimer’s Dement. 2024, 20, 4212–4233. [Google Scholar] [CrossRef]
- Wang, H.; Fu, J.; Xu, X.; Yang, Z.; Zhang, T. Rapamycin Activates Mitophagy and Alleviates Cognitive and Synaptic Plasticity Deficits in a Mouse Model of Alzheimer’s Disease. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 1707–1713. [Google Scholar] [CrossRef]
- Mito, T.; Vincent, A.E.; Faitg, J.; Taylor, R.W.; Khan, N.A.; McWilliams, T.G.; Suomalainen, A. Mosaic dysfunction of mitophagy in mitochondrial muscle disease. Cell Metab. 2022, 34, 197–208.e5. [Google Scholar] [CrossRef]
- Zheng, W.; Li, K.; Zhong, M.; Wu, K.; Zhou, L.; Huang, J.; Liu, L.; Chen, Z. Mitophagy activation by rapamycin enhances mitochondrial function and cognition in 5×FAD mice. Behav. Brain Res. 2024, 463, 114889. [Google Scholar] [CrossRef]
- Hofer, S.J.; Liang, Y.; Zimmermann, A.; Schroeder, S.; Dengjel, J.; Kroemer, G.; Eisenberg, T.; Sigrist, S.J.; Madeo, F. Spermidine-induced hypusination preserves mitochondrial and cognitive function during aging. Autophagy 2021, 17, 2037–2039. [Google Scholar] [CrossRef]
- Fairley, L.H.; Lejri, I.; Grimm, A.; Eckert, A. Spermidine Rescues Bioenergetic and Mitophagy Deficits Induced by Disease-Associated Tau Protein. Int. J. Mol. Sci. 2023, 24, 5297. [Google Scholar] [CrossRef]
- Liu, X.; Ye, M.; Ma, L. The emerging role of autophagy and mitophagy in tauopathies: From pathogenesis to translational implications in Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1022821. [Google Scholar] [CrossRef]
- Sigrist, S.J.; Carmona-Gutierrez, D.; Gupta, V.K.; Bhukel, A.; Mertel, S.; Eisenberg, T.; Madeo, F. Spermidine-triggered autophagy ameliorates memory during aging. Autophagy 2014, 10, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Schwarz, C.; Benson, G.; Horn, N.; Buchert, R.; Lange, C.; Köbe, T.; Hetzer, S.; Maglione, M.; Michael, E.; et al. Effects of spermidine supplementation on cognition and biomarkers in older adults with subjective cognitive decline (SmartAge)-study protocol for a randomized controlled trial. Alzheimer’s Res. Ther. 2019, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, S.; Cetrullo, S.; Guidotti, S.; Silvestri, Y.; Minguzzi, M.; Santi, S.; Cattini, L.; Filardo, G.; Flamigni, F.; Borzì, R.M. Spermidine rescues the deregulated autophagic response to oxidative stress of osteoarthritic chondrocytes. Free Radic. Biol. Med. 2020, 153, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Qiu, Q.; Gu, X.; Tian, Y.; Zhang, Y. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci. Rep. 2016, 6, 24700. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Gioscia-Ryan, R.A.; Hearon, C.M.; Seals, D.R. The autophagy enhancer spermidine reverses arterial aging. Mech. Ageing Dev. 2013, 134, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Sacitharan, P.K.; Lwin, S.; Gharios, G.B.; Edwards, J.R. Spermidine restores dysregulated autophagy and polyamine synthesis in aged and osteoarthritic chondrocytes via EP300. Exp. Mol. Med. 2018, 50, 1–10. [Google Scholar] [CrossRef]
- Morselli, E.; Mariño, G.; Bennetzen, M.V.; Eisenberg, T.; Megalou, E.; Schroeder, S.; Cabrera, S.; Bénit, P.; Rustin, P.; Criollo, A.; et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol. 2011, 192, 615–629. [Google Scholar] [CrossRef]
- Yue, F.; Li, W.; Zou, J.; Jiang, X.; Xu, G.; Huang, H.; Liu, L. Spermidine Prolongs Lifespan and Prevents Liver Fibrosis and Hepatocellular Carcinoma by Activating MAP1S-Mediated Autophagy. Cancer Res. 2017, 77, 2938–2951. [Google Scholar] [CrossRef]
- Yan, J.; Yan, J.-Y.; Wang, Y.-X.; Ling, Y.-N.; Song, X.-D.; Wang, S.-Y.; Liu, H.-Q.; Liu, Q.-C.; Zhang, Y.; Yang, P.-Z.; et al. Spermidine-enhanced autophagic flux improves cardiac dysfunction following myocardial infarction by targeting the AMPK/mTOR signalling pathway. Br. J. Pharmacol. 2019, 176, 3126–3142. [Google Scholar] [CrossRef]
- Pietrocola, F.; Lachkar, S.; Enot, D.P.; Niso-Santano, M.; Bravo-San Pedro, J.M.; Sica, V.; Izzo, V.; Maiuri, M.C.; Madeo, F.; Mariño, G.; et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015, 22, 509–516. [Google Scholar] [CrossRef]
- Xu, T.-T.; Li, H.; Dai, Z.; Lau, G.K.; Li, B.-Y.; Zhu, W.-L.; Liu, X.-Q.; Liu, H.-F.; Cai, W.-W.; Huang, S.-Q.; et al. Spermidine and spermine delay brain aging by inducing autophagy in SAMP8 mice. Aging 2020, 12, 6401–6414. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Yang, X.; Li, J.; Shu, Z.; Dai, J.; Liu, X.; Li, B.; Jia, S.; Kou, X.; Yang, Y.; et al. Spermidine coupled with exercise rescues skeletal muscle atrophy from D-gal-induced aging rats through enhanced autophagy and reduced apoptosis via AMPK-FOXO3a signal pathway. Oncotarget 2017, 8, 17475–17490. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.-H.; Yan, J.-L.; Wang, Q.-J.; Chen, H.-C.; Ma, X.-Z.; Yin, J.; Gao, L.-P. Spermidine ameliorates the neuronal aging by improving the mitochondrial function in vitro. Exp. Gerontol. 2018, 108, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Sharma, S.; Kumar, P.; Deshmukh, R. Neuroprotective potential of spermidine against rotenone induced Parkinson’s disease in rats. Neurochem. Int. 2018, 116, 104–111. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, M.; Hou, X.-O.; Hu, L.-F. Roles of microglial mitophagy in neurological disorders. Front. Aging Neurosci. 2022, 14, 979869. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Sawa-Makarska, J.; Khuu, G.; Lam, W.K.; Adriaenssens, E.; Fracchiolla, D.; Shoebridge, S.; Bernklau, D.; Padman, B.S.; Skulsuppaisarn, M.; et al. Unconventional initiation of PINK1/Parkin mitophagy by Optineurin. Mol. Cell 2023, 83, 1693–1709.e9. [Google Scholar] [CrossRef]
- Soh, J.E.C.; Shimizu, A.; Molla, M.R.; Zankov, D.P.; Nguyen, L.K.C.; Khan, M.R.; Tesega, W.W.; Chen, S.; Tojo, M.; Ito, Y.; et al. RhoA rescues cardiac senescence by regulating Parkin-mediated mitophagy. J. Biol. Chem. 2023, 299, 102993. [Google Scholar] [CrossRef]
- Zeviani, M.; Tiranti, V.; Piantadosi, C. Mitochondrial disorders. Medicine 1998, 77, 59–72. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Kang, M.-J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-G.; Miao, C.-Y. Mitochondrial transplantation as a promising therapy for mitochondrial diseases. Acta Pharm. Sin. B 2023, 13, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Q.; Xiang, L.; Chen, Y. Mitochondrial transplantation: A promising therapy for mitochondrial disorders. Int. J. Pharm. 2024, 658, 124194. [Google Scholar] [CrossRef]
- Hosseinian, S.; Ali Pour, P.; Kheradvar, A. Prospects of mitochondrial transplantation in clinical medicine: Aspirations and challenges. Mitochondrion 2022, 65, 33–44. [Google Scholar] [CrossRef]
- Gollihue, J.L.; Patel, S.P.; Rabchevsky, A.G. Mitochondrial transplantation strategies as potential therapeutics for central nervous system trauma. Neural Regen. Res. 2018, 13, 194–197. [Google Scholar] [CrossRef]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef]
- Glytsou, C.; Chen, X.; Zacharioudakis, E.; Al-Santli, W.; Zhou, H.; Nadorp, B.; Lee, S.; Lasry, A.; Sun, Z.; Papaioannou, D.; et al. Mitophagy Promotes Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Discov. 2023, 13, 1656–1677. [Google Scholar] [CrossRef]
- Elcocks, H.; Brazel, A.J.; McCarron, K.R.; Kaulich, M.; Husnjak, K.; Mortiboys, H.; Clague, M.J.; Urbé, S. FBXL4 ubiquitin ligase deficiency promotes mitophagy by elevating NIX levels. EMBO J. 2023, 42, e112799. [Google Scholar] [CrossRef]
- Davinelli, S.; De Stefani, D.; De Vivo, I.; Scapagnini, G. Polyphenols as Caloric Restriction Mimetics Regulating Mitochondrial Biogenesis and Mitophagy. Trends Endocrinol. Metab. 2020, 31, 536–550. [Google Scholar] [CrossRef]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef] [PubMed]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and mitochondrial health. J. Physiol. 2021, 599, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.C.; Wilson, R.J.; Laker, R.C.; Guan, Y.; Spaulding, H.R.; Nichenko, A.S.; Shen, W.; Shang, H.; Dorn, M.V.; Huang, K.; et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2025932118. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Suzuki, K.; Higuchi, M.; Balogh, L.; Boldogh, I.; Koltai, E. Physical exercise, reactive oxygen species and neuroprotection. Free Radic. Biol. Med. 2016, 98, 187–196. [Google Scholar] [CrossRef]
- Bernardo, T.C.; Marques-Aleixo, I.; Beleza, J.; Oliveira, P.J.; Ascensão, A.; Magalhães, J. Physical Exercise and Brain Mitochondrial Fitness: The Possible Role Against Alzheimer’s Disease. Brain Pathol. 2016, 26, 648–663. [Google Scholar] [CrossRef]
- Dietrich, M.O.; Andrews, Z.B.; Horvath, T.L. Exercise-induced synaptogenesis in the hippocampus is dependent on UCP2-regulated mitochondrial adaptation. J. Neurosci. 2008, 28, 10766–10771. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Callio, J.; Distefano, G.; O’Doherty, R.M.; Goodpaster, B.H.; Coen, P.M.; Chu, C.T. Exercise increases mitochondrial complex I activity and DRP1 expression in the brains of aged mice. Exp. Gerontol. 2017, 90, 1–13. [Google Scholar] [CrossRef]
- Safdar, A.; Saleem, A.; Tarnopolsky, M.A. The potential of endurance exercise-derived exosomes to treat metabolic diseases. Nat. Rev. Endocrinol. 2016, 12, 504–517. [Google Scholar] [CrossRef]
- Giudice, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Covington, J.D.; Tam, C.S.; Bajpeyi, S.; Galgani, J.E.; Noland, R.C.; Smith, S.R.; Redman, L.M.; Ravussin, E. Myokine Expression in Muscle and Myotubes in Response to Exercise Stimulation. Med. Sci. Sports Exerc. 2016, 48, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.L.P.; Fernandes, T.; Soci, U.P.R.; Silveira, A.C.; Barretti, D.L.M.; Negrão, C.E.; Oliveira, E.M. Obesity Downregulates MicroRNA-126 Inducing Capillary Rarefaction in Skeletal Muscle: Effects of Aerobic Exercise Training. Oxid. Med. Cell. Longev. 2017, 2017, 2415246. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Song, W. Resistance training increases fibroblast growth factor-21 and irisin levels in the skeletal muscle of Zucker diabetic fatty rats. J. Exerc. Nutr. Biochem. 2017, 21, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Dzah, C.S.; Asante-Donyinah, D.; Letsyo, E.; Dzikunoo, J.; Adams, Z.S. Dietary Polyphenols and Obesity: A Review of Polyphenol Effects on Lipid and Glucose Metabolism, Mitochondrial Homeostasis, and Starch Digestibility and Absorption. Plant Foods Hum. Nutr. 2023, 78, 1–12. [Google Scholar] [CrossRef]
- Yoshida, Y.; Tamura, Y.; Kouzaki, K.; Nakazato, K. Dietary apple polyphenols enhance mitochondrial turnover and respiratory chain enzymes. Exp. Physiol. 2023, 108, 1295–1307. [Google Scholar] [CrossRef]
- Gureev, A.P.; Andrianova, N.V.; Pevzner, I.B.; Zorova, L.D.; Chernyshova, E.V.; Sadovnikova, I.S.; Chistyakov, D.V.; Popkov, V.A.; Semenovich, D.S.; Babenko, V.A.; et al. Dietary restriction modulates mitochondrial DNA damage and oxylipin profile in aged rats. FEBS J. 2022, 289, 5697–5713. [Google Scholar] [CrossRef]
- Waldman, M.; Cohen, K.; Yadin, D.; Nudelman, V.; Gorfil, D.; Laniado-Schwartzman, M.; Kornwoski, R.; Aravot, D.; Abraham, N.G.; Arad, M.; et al. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving “SIRT1 and PGC-1α”. Cardiovasc. Diabetol. 2018, 17, 111. [Google Scholar] [CrossRef]
- Davinelli, S.; Sapere, N.; Visentin, M.; Zella, D.; Scapagnini, G. Enhancement of mitochondrial biogenesis with polyphenols: Combined effects of resveratrol and equol in human endothelial cells. Immun. Ageing 2013, 10, 28. [Google Scholar] [CrossRef]
- Han, X.; Xu, T.; Fang, Q.; Zhang, H.; Yue, L.; Hu, G.; Sun, L. Quercetin hinders microglial activation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy. Redox Biol. 2021, 44, 102010. [Google Scholar] [CrossRef]
- Dolman, N.J.; Chambers, K.M.; Mandavilli, B.; Batchelor, R.H.; Janes, M.S. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy 2013, 9, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Berezhnov, A.V.; Soutar, M.P.M.; Fedotova, E.I.; Frolova, M.S.; Plun-Favreau, H.; Zinchenko, V.P.; Abramov, A.Y. Intracellular pH Modulates Autophagy and Mitophagy. J. Biol. Chem. 2016, 291, 8701–8708. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Rosado, C.J.; Mijaljica, D.; Hatzinisiriou, I.; Prescott, M.; Devenish, R.J. Rosella: A fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy 2008, 4, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; Andrabi, S.A.; Chen, W.; Höke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014, 33, 2798–2813. [Google Scholar] [CrossRef]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef]
- Zhuo, Z.; Lin, H.; Liang, J.; Ma, P.; Li, J.; Huang, L.; Chen, L.; Yang, H.; Bai, Y.; Sha, W. Mitophagy-Related Gene Signature for Prediction Prognosis, Immune Scenery, Mutation, and Chemotherapy Response in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 802528. [Google Scholar] [CrossRef]
- Yang, M.; Fu, J.-D.; Zou, J.; Sridharan, D.; Zhao, M.-T.; Singh, H.; Krigman, J.; Khan, M.; Xin, G.; Sun, N. Assessment of mitophagy in human iPSC-derived cardiomyocytes. Autophagy 2022, 18, 2481–2494. [Google Scholar] [CrossRef]
- Caponio, D.; Veverová, K.; Zhang, S.; Shi, L.; Wong, G.; Vyhnalek, M.; Fang, E.F. Compromised autophagy and mitophagy in brain ageing and Alzheimer’s diseases. Aging Brain 2022, 2, 100056. [Google Scholar] [CrossRef] [PubMed]
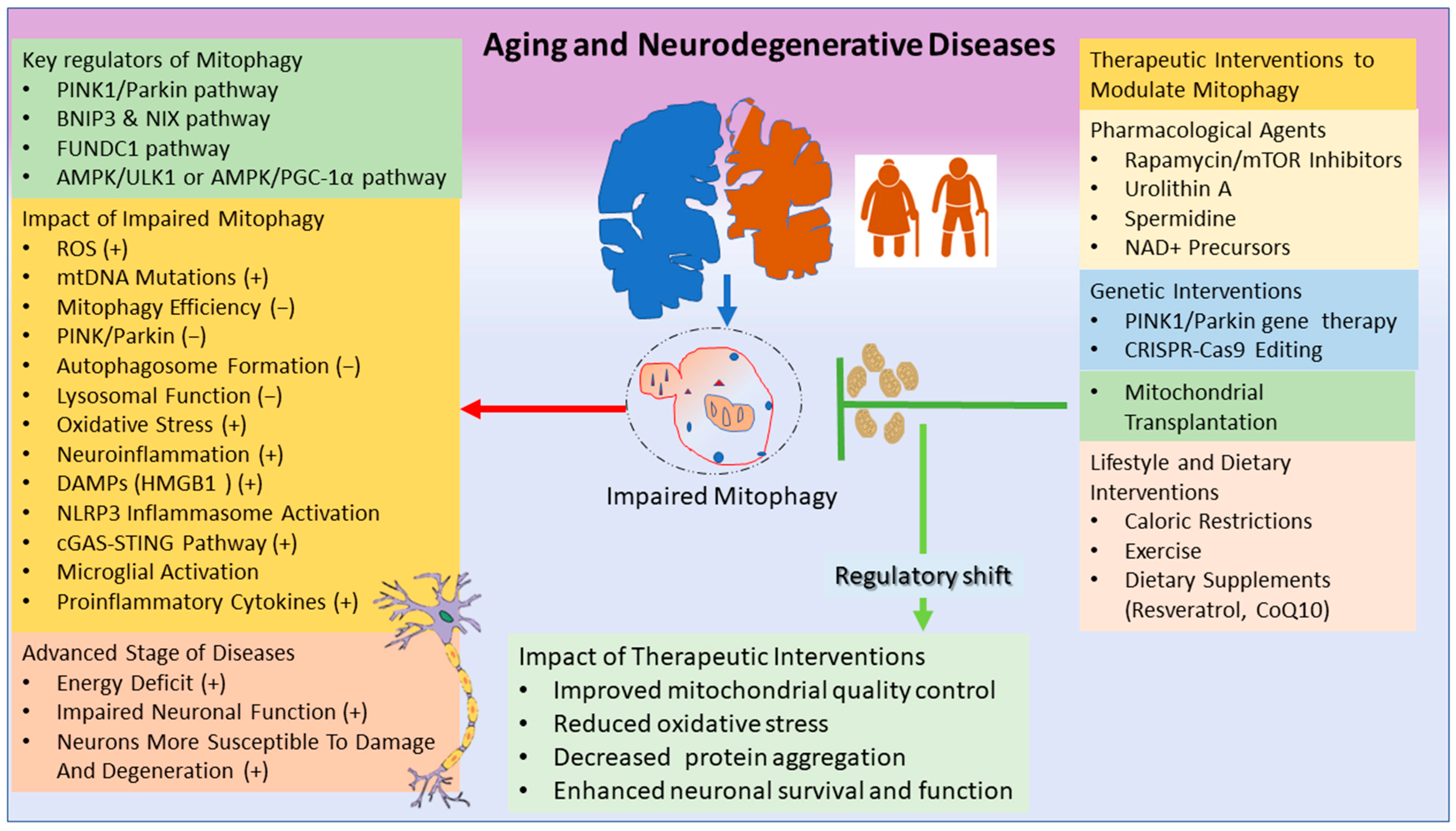

{kind=link}
| Regulator | Function | Pathway | Relevance to Disease | References |
|---|---|---|---|---|
| PINK1 | Accumulates on the outer mitochondrial membrane upon depolarization, recruits Parkin | PINK1/Parkin Pathway | Impaired function linked to Parkinson’s Disease | [4,16,17,18,19] |
| Parkin | Ubiquitinates mitochondrial surface proteins, marking them for degradation | PINK1/Parkin Pathway | Mutations associated with familial Parkinson’s Disease | [4,16,17,18,19,20] |
| BNIP3 | Interacts with LC3 to promote mitophagy under hypoxia | BNIP3 Pathway | Involved in hypoxia-induced mitophagy in cancer and cardiac tissues | [21,22] |
| NIX | Similar to BNIP3, interacts with LC3 during hypoxia | BNIP3 Pathway | Important for erythrocyte maturation and heart function | [21,22] |
| FUNDC1 | Mediates hypoxia-induced mitophagy through interaction with LC3 | FUNDC1 Pathway | Plays a role in ischemic heart diseases | [23,24] |
| AMPK | Initiates mitophagy by phosphorylating ULK1(vital for neuroprotection). Also inhibits mTORC1. | AMPK-ULK1 pathway | Reduces neuroinflammation and protects neuronal cells. | [25,26,27] |
| Disease | Mitophagy Impairment | Molecular Markers | Therapeutic Approaches | References |
|---|---|---|---|---|
| Alzheimer’s Disease | Reduced PINK1/Parkin activity, accumulation of damaged mitochondria | Decreased PINK1, Parkin levels | Urolithin A, Rapamycin, lifestyle interventions (exercise, caloric restriction) | [8,12,18] |
| Parkinson’s Disease | Mutations in PINK1, Parkin lead to impaired mitophagy | Reduced Parkin-mediated ubiquitination | Gene therapy (PINK1/Parkin), mitochondrial transplantation | [2,9,19,60] |
| Huntington’s Disease | Accumulation of damaged mitochondria due to impaired mitophagy | Altered mitochondrial dynamics proteins (e.g., Drp1) | Pharmacological agents, lifestyle interventions | [12] |
| Multiple Sclerosis | Accumulation of damaged mitochondria due to impaired mitophagy | Altered mitochondrial dynamics proteins (e.g., Drp1) | Pharmacological agents, lifestyle interventions | [62] |
| Agent | Mechanism of Action | Evidence from Studies | Potential Therapeutic Use | References |
|---|---|---|---|---|
| Urolithin A | Induces mitophagy, promotes mitochondrial health, stimulates mitochondrial biogenesis. | Improves mitochondrial function, muscle function, and lifespan; enhances cognitive functions, synaptic plasticity; reduces neuroinflammation and neuron loss in neurodegenerative models. | Neurodegenerative diseases (e.g., Alzheimer’s, Parkinson’s), Aging. | [9,63,64] |
| Rapamycin | Inhibits mTOR, enhances autophagy and mitophagy, aids in clearance of damaged mitochondria. | Improves learning, memory, synaptic plasticity, and mitochondrial function; reduces oxidative stress, apoptosis, and neuronal loss in neurodegenerative models. | Alzheimer’s Disease, longevity | [10,65,66,67] |
| Spermidine | Stimulates autophagy and mitophagy (markers, including Beclin-1, LC3-II, PINK1, PARKIN, ULK1, Atg, AMPK, and inhibiting mTOR), aids in the removal of dysfunctional mitochondria. | Enhances cognitive function, decreases oxidative stress, extends lifespan; improves memory performance in older adults | Improves cognitive decline, promotes neuroprotection. | [11,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86] |
| NAD+ Precursors | Increases intracellular NAD+ levels, activates sirtuins (e.g., SIRT1, SIRT3) involved in mitochondrial biogenesis and quality control. | Restores mitochondrial function, reduces oxidative stress in astrocytes and microglia. | Neurodegenerative diseases, mitochondrial dysfunction. | [57,87] |
| PINK1 and Parkin Gene Therapy | Restores mitophagy by introducing functional copies of PINK1 or Parkin genes. | Corrects mitochondrial defects, reduces neuroinflammation, enhances motor function in Parkinson’s disease models. | Parkinson’s disease, other neurodegenerative diseases. | [88,89] |
| Mitochondrial Transplantation | Transplants healthy mitochondria into cells with dysfunctional ones. | Enhances cellular function and alleviates symptoms of neurodegenerative diseases. | Neurodegenerative diseases characterized by mitochondrial dysfunction. | [90,91,92,93,94,95,96] |
| CRISPR/Cas9-Based Therapies | Edits genes to correct mutations disrupting mitophagy, restores normal mitochondrial function. | Ongoing research focused on mitochondrial myopathies and neurodegenerative diseases. | Mitochondrial myopathies, neurodegenerative diseases. | [97,98,99] |
| Caloric Restriction (CR) | Enhances mitophagy, improves mitochondrial function, reduces oxidative stress and reduces inflammatory markers | Extends lifespan, delays age-related diseases, improves metabolic health and cognitive function. Decreases fasting insulin levels, body temperature, resting energy expenditure and thyroid axis activity | Aging, cardiometabolic risk, metabolic health, cognitive function. | [100,101] |
| Exercise | Promotes elimination of damaged mitochondria, stimulates mitochondrial biogenesis. | Improves cognitive function, therapeutic strategy for dementia patients, delays neurodegenerative diseases, increases angiogenesis, neurogenesis, reducing age-related brain atrophy and supports healthy aging. | Neurodegenerative diseases, healthy aging. | [102,103,104,105,106,107,108,109,110,111,112,113,114] |
| Dietary Polyphenols | Enhances mitophagy, reduces oxidative stress, improves mitochondrial function. | Improves cognitive function, reduces inflammation and oxidative stress in neurodegenerative models. | Aging, neurodegenerative diseases. | [80,100,115,116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, D.; Kumar, A. Harnessing Mitophagy for Therapeutic Advances in Aging and Chronic Neurodegenerative Diseases. Neuroglia 2024, 5, 391-409. https://doi.org/10.3390/neuroglia5040026
Ghosh D, Kumar A. Harnessing Mitophagy for Therapeutic Advances in Aging and Chronic Neurodegenerative Diseases. Neuroglia. 2024; 5(4):391-409. https://doi.org/10.3390/neuroglia5040026
Chicago/Turabian StyleGhosh, Devlina, and Alok Kumar. 2024. "Harnessing Mitophagy for Therapeutic Advances in Aging and Chronic Neurodegenerative Diseases" Neuroglia 5, no. 4: 391-409. https://doi.org/10.3390/neuroglia5040026
APA StyleGhosh, D., & Kumar, A. (2024). Harnessing Mitophagy for Therapeutic Advances in Aging and Chronic Neurodegenerative Diseases. Neuroglia, 5(4), 391-409. https://doi.org/10.3390/neuroglia5040026