The Influences of Magnesium upon Calcium Phosphate Mineral Formation and Structure as Monitored by X-ray and Vibrational Spectroscopy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mineral Synthesis
2.1.1. Brushite (CaHPO4·2H2O) and Related Mixed Species Synthesis
2.1.2. Newberyite (MgHPO4·3H2O) and Related Mixed Species Synthesis
2.1.3. Hydroxyapatite (Ca5(PO4)3(OH)) and Related Mixed Species Synthesis
2.1.4. Bobierrite (Mg3(PO4)2·8H2O) and Related Mixed Species Synthesis
2.2. Synchrotron Powder X-Ray Diffraction
2.3. Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) Spectroscopy
2.4. Elemental Analysis
2.5. Transmission Electron Microscopy (TEM)
2.6. Phosphorus K-Edge XANES Spectroscopy
3. Results
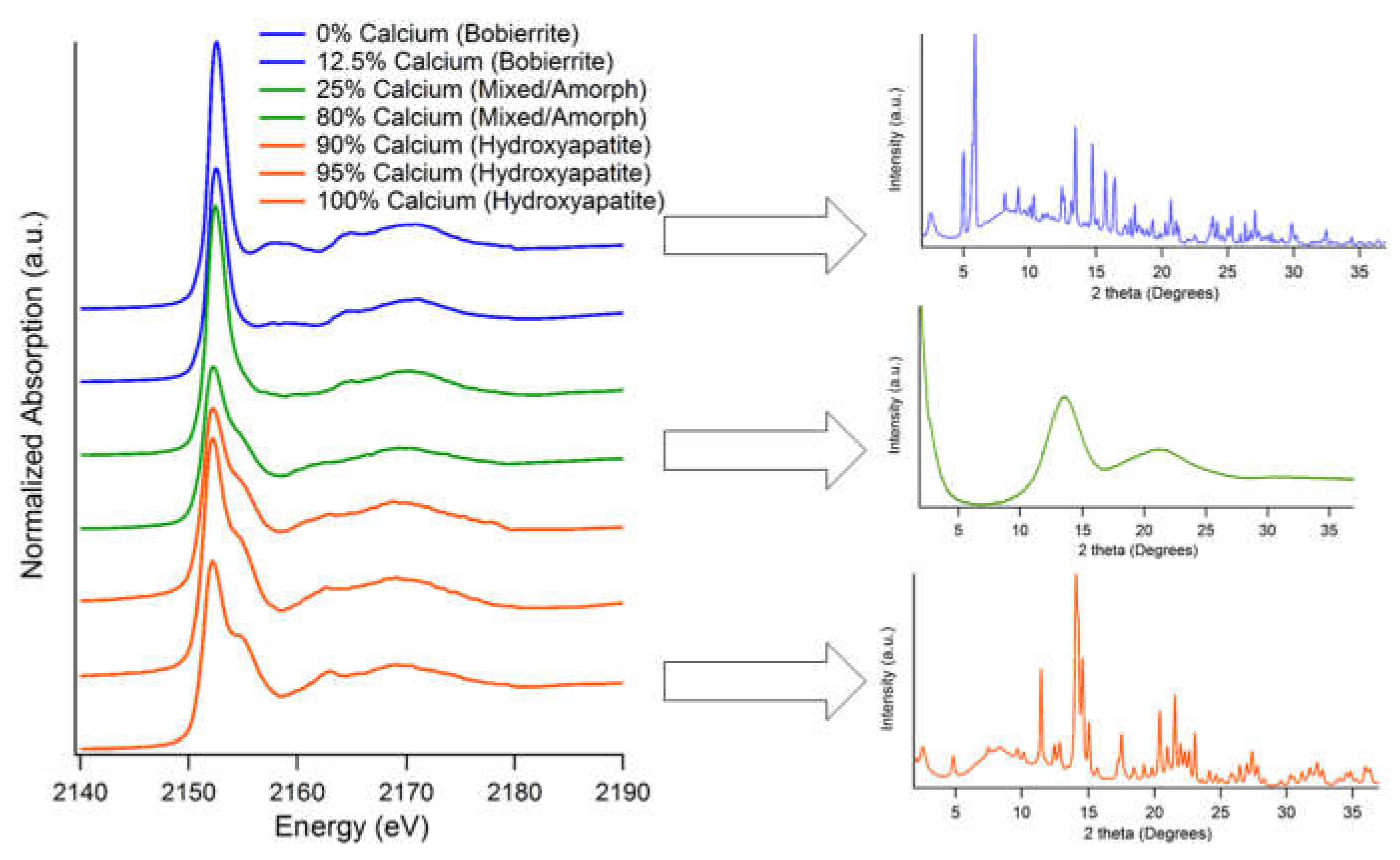
Hydroxyapatite and Bobierrite Mineral Series
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dorozhkin, S.V. Calcium Orthophosphates: Occurrence, Properties, Biomineralization, Pathological Calcification and Biomimetic Applications. Biomatter 2011, 1, 121–164. [Google Scholar] [CrossRef] [PubMed]
- Kar, G.; Hundal, L.S.; Schoenau, J.J.; Peak, D. Direct Chemical Speciation of P in Sequential Chemical Extraction Residues Using P K-Edge X-Ray Absorption near-Edge Structure Spectroscopy. Soil Sci. 2011, 176, 589–595. [Google Scholar] [CrossRef]
- Peak, D.; Kar, G.; Hundal, L.; Schoenau, J. Kinetics and Mechanisms of Phosphorus Release in a Soil Amended With Biosolids or Inorganic Fertilizer. Soil Sci. 2012, 177, 183–187. [Google Scholar] [CrossRef]
- Manimel Wadu, M.C.W.; Michaelis, V.K.; Kroeker, S.; Akinremi, O.O. Exchangeable Calcium/Magnesium Ratio Affects Phosphorus Behavior in Calcareous Soils. Soil Sci. Soc. Am. J. 2013, 77, 2004. [Google Scholar] [CrossRef]
- Johnsson, M.S.; Nancollas, G.H. The Role of Brushite and Octacalcium Phosphate in Apatite Formation. Crit. Rev. Oral Biol. Med. 1992, 3, 61–82. [Google Scholar] [CrossRef]
- Valsami-Jones, E. Mineralogical Controls on Phosphorus Recovery from Wastewaters. Mineral. Mag. 2001, 65, 611–620. [Google Scholar] [CrossRef]
- Racz, G.; Soper, R. Reaction Products of Orthophosphates in Soils Containing Varying Amounts of Calcium and Magnesium. Can. J. Soil Sci. 1967, 47, 223–230. [Google Scholar] [CrossRef]
- Bauge, S.M.Y.; Lavkulich, L.M.; Schreier, H.E. Serpentine Affected Soils and the Formation of Magnesium Phosphates (Struvite). Can. J. Soil Sci. 2013, 93, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.; Adam, C.; Sekine, R.; Schiller, T.; Lipie, E.; McNaughton, D. Determination of Phosphorus Fertilizer Soil Reactions by Raman and Synchrotron Infrared Microspectroscopy. Appl. Spectrosc. 2013, 67, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Siciliano, S.D.; Chen, T.; Phillips, C.; Hamilton, J.; Hilger, D.; Chartrand, B.; Grosskleg, J.; Bradshaw, K.; Carlson, T.; Peak, D. Total Phosphate Influences the Rate of Hydrocarbon Degradation but Phosphate Mineralogy Shapes Microbial Community Composition in Cold-Region Calcareous Soils. Environ. Sci. Technol. 2016, 50, 5197–5206. [Google Scholar] [CrossRef]
- Salimi, M.H.; Heughebaert, J.C.; Nancollas, G.H. Crystal Growth of Calcium Phosphates in the Presence of Magnesium Ions. Langmuir 1985, 1, 119–122. [Google Scholar] [CrossRef]
- Wang, L.; Nancollas, G.H. Calcium Orthophosphates: Crystallization and Dissolution. Chem. Rev. 2008, 108, 4628–4669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Harris, W. Carbonate and Magnesium Interactive Effect on Calcium Phosphate Precipitation. Environ. Sci. Technol. 2008, 42, 436–442. [Google Scholar] [CrossRef]
- Boanini, E.; Gazzano, M.; Bigi, A. Ionic Substitutions in Calcium Phosphates Synthesized at Low Temperature. Acta Biomater. 2010, 6, 1882–1894. [Google Scholar] [CrossRef]
- Diallo-Garcia, S.; Laurencin, D.; Krafft, J.M.; Casale, S.; Smith, M.E.; Lauron-Pernot, H.; Costentin, G. Influence of Magnesium Substitution on the Basic Properties of Hydroxyapatites. J. Phys. Chem. C 2011, 115, 24317–24327. [Google Scholar] [CrossRef]
- Driessens, F.C.M.; Verbeek, R.M.H. The Calcium Rich Compounds of the System Ca(OH)2-H3PO4-H2O; CRC Press: Boca Raton, FL, USA, 1990; pp. 37–59. [Google Scholar]
- Cao, X.; Harris, W.G.; Josan, M.S.; Nair, V.D. Inhibition of Calcium Phosphate Precipitation under Environmentally-Relevant Conditions. Sci. Total Environ. 2007, 383, 205–215. [Google Scholar] [CrossRef]
- Hesterberg, D. Chapter 11 – Macroscale Chemical Properties and X-Ray Absorption Spectroscopy of Soil Phosphorus. Dev. Soil Sci. 2010, 34, 313–356. [Google Scholar] [CrossRef]
- Franke, R.; Hormes, J. The P K-near Edge Absorption Spectra of Phosphates. Phys. B Phys. Condens. Matter 1995, 216, 85–95. [Google Scholar] [CrossRef]
- Hesterberg, D.; Zhou, W.; Hutchison, K.J.; Beauchemin, S.; Sayers, D.E. XAFS Study of Adsorbed and Mineral Forms of Phosphate. J. Synchrotron Radiat. 1999, 6, 636–638. [Google Scholar] [CrossRef]
- Okude, N.; Nagoshi, M.; Noro, H.; Baba, Y.; Yamamoto, H.; Sasaki, T. P and S K-Edge XANES of Transition-Metal Phosphates and Sulfates. J. Electron Spectros. Relat. Phenomena 1999, 101–103, 607–610. [Google Scholar] [CrossRef]
- Werner, F.; Prietzel, J. Standard Protocol and Quality Assessment of Soil Phosphorus Speciation by P K-Edge XANES Spectroscopy. Environ. Sci. Technol. 2015, 49, 10521–10528. [Google Scholar] [CrossRef] [PubMed]
- Peak, D.; Sims, J.T.; Sparks, D.L. Solid-State Speciation of Natural and Alum-Amended Poultry Litter Using XANES Spectroscopy. Environ. Sci. Technol. 2002, 36, 4253–4261. [Google Scholar] [CrossRef] [PubMed]
- Ajiboye, B.; Akinremi, O.O.; Hu, Y.; Jürgensen, A. XANES Speciation of Phosphorus in Organically Amended and Fertilized Vertisol and Mollisol. Soil Sci. Soc. Am. J. 2008, 72, 1256. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J.; Cade-Menun, B.J.; Liang, X.; Hu, Y.; Liu, C.W.; Zhao, Y.; Li, L.; Shi, J. Complementary Phosphorus Speciation in Agricultural Soils by Sequential Fractionation, Solution P Nuclear Magnetic Resonance, and Phosphorus K-Edge X-Ray Absorption Near-Edge Structure Spectroscopy. J. Environ. Qual. 2013, 42, 1763. [Google Scholar] [CrossRef]
- Kar, G.; Schoenau, J.; Hilger, D.; Peak, D. Direct Chemical Speciation of Soil Phosphorus in a Saskatchewan Chernozem after Long and Short-Term Manure Amendments. Can. J. Soil Sci. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Ingall, E.D.; Brandes, J.A.; Diaz, J.M.; De Jonge, M.D.; Paterson, D.; McNulty, I.; Elliott, W.C.; Northrup, P. Phosphorus K-Edge XANES Spectroscopy of Mineral Standards. J. Synchrotron Radiat. 2011, 18, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Oxmann, J.F. Technical Note: An X-Ray Absorption Method for the Identification of Calcium Phosphate Species Using Peak-Height Ratios. Biogeosciences 2014, 11, 2169–2183. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Kumta, P.N. Chemical Synthesis and Characterization of Magnesium Substituted Amorphous Calcium Phosphate (MG-ACP). Mater. Sci. Eng. C 2010, 30, 1313–1317. [Google Scholar] [CrossRef]
- Aramendía, M.A.; Borau, V.; Jiménez, C.; Marinas, J.M.; Romero, F.J. Synthesis and Characterization of Magnesium Phosphates and Their Catalytic Properties in the Conversion of 2-Hexanol. J. Colloid Interface Sci. 1999, 217, 288–298. [Google Scholar]
- Riman, R.E.; Sever, C. Biomimetic Hydroxyapatite Synthesis. US. Patent 8287914 B2, 16 October 2012. [Google Scholar]
- Laurencin, D.; Almora-Barrios, N.; de Leeuw, N.H.; Gervais, C.; Bonhomme, C.; Mauri, F.; Chrzanowski, W.; Knowles, J.C.; Newport, R.J.; Wong, A.; et al. Magnesium Incorporation into Hydroxyapatite. Biomaterials 2011, 32, 1826–1837. [Google Scholar] [CrossRef] [Green Version]
- Kolmas, J.; Jaklewicz, A.; Zima, A.; Bućko, M.; Paszkiewicz, Z.; Lis, J.; Ślósarczyk, A.; Kolodziejski, W. Incorporation of Carbonate and Magnesium Ions into Synthetic Hydroxyapatite: The Effect on Physicochemical Properties. J. Mol. Struct. 2011, 987, 40–50. [Google Scholar] [CrossRef]
- Toby, B.H.; Von Dreele, R.B. GSAS-II: The Genesis of a Modern Open-Source All Purpose Crystallography Software Package. J. Appl. Crystallogr. 2013, 46, 544–549. [Google Scholar] [CrossRef]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The HighScore Suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef] [Green Version]
- Larson, A.C.; Von Dreele, R.B. General Structure Analysis System; report LAUR 86-748; Los Alamos National Laboratory: Los Alamos, NM, USA, 2004. [Google Scholar] [CrossRef]
- Schofield, P.F.; Knight, K.S.; van der Houwen, J.A.M.; Valsami-Jones, E. The Role of Hydrogen Bonding in the Thermal Expansion and Dehydration of Brushite, Di-Calcium Phosphate Dihydrate. Phys. Chem. Miner. 2004, 31, 606–624. [Google Scholar] [CrossRef]
- Waseda, Y.; Matsubara, E.; Shinoda, K. X-Ray Diffraction Crystallography; Springer Science & Business Media: Berlin, Germany, 2011. [Google Scholar] [CrossRef]
- Stipniece, L.; Salma-Ancane, K.; Jakovlevs, D.; Borodajenko, N.; Berzina-Cimdina, L. The Study of Magnesium Substitution Effect on Physicochemical Properties of Hydroxyapatite. Mater. Sci. Appl. Chem. 2013, 28, 51. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, W.L. Chemical Equilibria in Soils; John Wiley & Sons: Chichester, UK, 1979. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Final Solid Composition (Ca:Mg) | |||||
|---|---|---|---|---|---|
| Acidic | Basic | ||||
| Initial Solution (Ca:Mg) | % Initial Ca | DCPD † | NBTE ‡ | HAP § | BOB ¶ |
| 1:0 | 100 | 1:0 | -- | 1:0 | -- |
| 20:1 | 95 | -- | -- | 1:0.02 | -- |
| 10:1 | 90 | -- | -- | 1:0.02 | -- |
| 5:1 | 80 | -- | -- | 1:0.06 | -- |
| 3:1 | 75 | 1:0.05 | -- | -- | -- |
| 1:1 | 50 | 1:0.35 | -- | -- | -- |
| 1:3 | 25 | -- | 0.7:1 | -- | 0.59:1 |
| 1:8 | 12.5 | -- | 0.35:1 | -- | 0.21:1 |
| 0:1 | 0 | -- | 0:1 | -- | 0:1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hilger, D.M.; Hamilton, J.G.; Peak, D. The Influences of Magnesium upon Calcium Phosphate Mineral Formation and Structure as Monitored by X-ray and Vibrational Spectroscopy. Soil Syst. 2020, 4, 8. https://doi.org/10.3390/soilsystems4010008
Hilger DM, Hamilton JG, Peak D. The Influences of Magnesium upon Calcium Phosphate Mineral Formation and Structure as Monitored by X-ray and Vibrational Spectroscopy. Soil Systems. 2020; 4(1):8. https://doi.org/10.3390/soilsystems4010008
Chicago/Turabian StyleHilger, David M., Jordan G. Hamilton, and Derek Peak. 2020. "The Influences of Magnesium upon Calcium Phosphate Mineral Formation and Structure as Monitored by X-ray and Vibrational Spectroscopy" Soil Systems 4, no. 1: 8. https://doi.org/10.3390/soilsystems4010008
APA StyleHilger, D. M., Hamilton, J. G., & Peak, D. (2020). The Influences of Magnesium upon Calcium Phosphate Mineral Formation and Structure as Monitored by X-ray and Vibrational Spectroscopy. Soil Systems, 4(1), 8. https://doi.org/10.3390/soilsystems4010008